
ХЄТЄЈєGµ°°ЧЕјБЄКЬМе(GPCRs)КЗІёИй¶ЇОпМеДЪЧоґуµДПё°ыД¤±нГжКЬМејТЧеЈ¬ѕЯУР7ґОїзД¤ВЭРэЅб№№Ј¬ИЛАа»щТтЧй±аВлФј800ЦЦІ»Н¬АаРНµДGPCRsЈ¬№г·єІОУлБЛґъР»РФјІІЎј°ЦЧБцµИ¶аЦЦЦШґујІІЎµДІЎАн№эіМЈ¬К№Ц®іЙОЄТ©ОпСР·ўµДИИГЕ°РµгЎЈлДКЗЅйУЪ°±»щЛбєНµ°°ЧЦКЦ®јдµДТ»АаОпЦКЈ¬УЙБЅёцЦБјёК®ёц°±»щЛбНЁ№элДјьБ¬ЅУ¶шіЙЈ¬КЗЙжј°ЙъОпМеДЪ¶аЦЦПё°ы№¦ДЬµДЙъОп»оРФОпЦКЎЈЖщЅсОЄЦ№Ј¬СРѕїХЯТСјш¶Ёіц7000УаЦЦМмИ»лДЈ¬·Ц±рЧчОЄј¤ЛШЎўЙсѕµЭЦКЎўЙъі¤ТтЧУЎўАлЧУНЁµАЕдМеєНї№ЙъЛШµИ·ў»У№¦ДЬЎЈлДАаТ©ОпТтѕЯУРЧчУГ»ъЦЖГчИ·Ўў°ІИ«РФєГЎўЙъІъіЙ±ѕµНµИ¶аЦШ¶АМШУЕКЖј°ЖдФЪїХјдЅб№№ЙПЅьєхОЮПЮµДїЙДЬРФ¶шЦрЅҐКЬµЅЦШКУЎЈЅьДкАґЈ¬»щУЪ¶ФGPCRЅб№№µДАнЅвІ»¶ПЙоИлЈ¬°РПтGPCRµДлДАаТ©Оп·ўХ№СёГНЈ¬РВТ©І»¶ПЙПКРЈ¬ДїЗ°ОЄЦ№Ј¬ГА№ъКіЖ·єНТ©Оп№ЬАнѕЦ(FDA)ТСЕъЧјЙПКРЅь50ЦЦ°РПтGPCRµДлДАаТ©ОпЈ¬УГУЪЦОБЖґъР»РФјІІЎЎўЙсѕПµНіјІІЎєН°©ЦўµИ¶аЦЦјІІЎЈ¬лДАаТ©ОпµДСР·ўѕАъБЛИЛМелДїЄ·ўЎўМмИ»лДїЄ·ўєНЙъОпјјКхїЄ·ўЈіёцЅЧ¶ОЎЈДїЗ°Ј¬ТСЙПКРµД°РПтGPCRлДАаТ©Опґу¶аКЗ¶ФИЛМеМмИ»ДЪФґРФ¶алДАаЕдМеµДёДФмУлРЮКОЈ¬±ѕОД№йДЙБЛЅьДкАґСР·ўіЙ№¦ТСЙПКРµД°РПтGPCRлДАаТ©ОпЈ¬ІўјтТЄЧЬЅбБЛДїЗ°лДАаТ©ОпµДСР·ўІЯВФј°ОґАґЗ±ФЪµД·ўХ№·ЅПтЈ¬ЦјФЪОЄёь¶а°РПтGPCRлДАаТ©ОпµДСР·ўМṩІОїјЎЈ
ЧчОЄИЛМеДЪЧоґуµДПё°ыД¤±нГжКЬМејТЧеЈ¬ЈЗµ°°ЧЕјБЄКЬМе(GPCRs)ІОУлБЛ°ьАЁМЗДтІЎЎўРДСЄ№ЬјІІЎЎўЧФЙнГвТЯІЎЎўЦЧБцµИФЪДЪµД¶аЦЦЦШґујІІЎµДІЎАн№эіМЎЈ°РПтGPCRТ©ОпµДїЄ·ўТ»Ц±КЗТ©ОпСР·ўµДИИµгЈ¬ГА№ъКіЖ·єНТ©Оп№ЬАнѕЦ(FDA) ЕъЧјµДИ«ІїТ©ОпЦРУРі¬№эЈ±/Јі¶јТФGPCRОЄЧчУðег[Ј±]ЎЈлДКЗЅйУЪ°±»щЛбєНµ°°ЧЦКЦ®јдµДТ»АаОпЦКЈ¬УЙЈІёцЦБјёК®ёц°±»щЛбНЁ№элДјьБ¬ЅУ¶шіЙЈ¬ЖщЅсОЄЦ№Ј¬ТСјш¶Ёіц7000УаЦЦМмИ»лДЈ¬·Ц±рЧчОЄј¤ЛШЎўЙсѕµЭЦКЎўЙъі¤ТтЧУЎўАлЧУНЁµАЕдМеєНї№ЙъЛШµИ·ў»У№¦ДЬ[ЈІ-Јі]Ј¬ФЪИЛМеДЪЈ¬лДАаІОУл¶аЦЦЦШТЄЙъАн№эіМ:АэИзТИµєЛШ(insulin)ґЩЅшМЗФЎўЦ¬·ѕєНµ°°ЧЦКµДєПіЙґъР»Ј¬·ў»УЅµСЄМЗµДЧчУГ[Јґ-Јµ]ЎЈУЦИзґЯІъЛШ(oxytocin)ФЪ·ЦГд№эіМЦРґЩЅшЧУ№¬ЖЅ»¬јЎµДКХЛхЈ¬ІўґМј¤ИйПЩЙдИй·ґУ¦[Ј¶]ЎЈлДЧчОЄТ»АаРЕєЕ·ЦЧУЈ¬НЁ№эЅбєППё°ыД¤±нГжКЬМеЈ¬АэИзGPCRЈуЎўАлЧУНЁµА(ion channels) µИЈ¬Жф¶ЇМШ¶ЁµДПё°ыРЕєЕЧЄµј№эіМЎЈЅьДкАґЈ¬лДАаТ©ОпЦрЅҐТэЖрТЅТ©ЅзµДЦШКУЈ¬ЖдЖѕЅиЙъОп»оРФёЯЎў°ІИ«РФєГЎўЙъІъіЙ±ѕµНµИМШРФј°ЖдФЪїХјдЅб№№ЙПЅьєхОЮПЮµДїЙДЬРФ±»КУОЄТ©ОпСР·ўµДРВФці¤µгЎЈФЪ±ѕЖЄЧЫКцЦРЈ¬№йДЙБЛСР·ўіЙ№¦ТСЙПКРµД°РПтGPCRлДАаТ©ОпЈ¬ІўјтТЄЧЬЅбБЛДїЗ°лДАаТ©ОпµДСР·ўІЯВФєНОґАґїЙДЬµД·ўХ№·ЅПтЎЈ
Ј±ЈЗµ°°ЧЕјБЄКЬМеј°ЖдРЕєЕЧЄµјНЁВ·
ИЛАа»щТтЧй±аВлі¬№э800ЦЦGPCRЈ¬ЖдЕдМеїЙТФКЗРЎ·ЦЧУМЗАаЎўЦ¬ЦКєН¶алДЈ¬ТІїЙТФКЗµ°°ЧЦКµИЙъОпґу·ЦЧУЈ¬GPCRµД№ІН¬МШµгКЗЖдБўМеЅб№№°ьє¬7ёцїзД¤ ¦Б ВЭРэЈ¬ТтґЛУЦ±»іЖОЄ,7ґОїзД¤КЬМеЈ¬Н¬К±Жд°ыДЪЗшУРGµ°°Ч(ДсЬХЛбЅбєПµ°°Ч)ЅбєПО»µг[Ј·]ЎЈёщѕЭЖдЅб№№µДПаЛЖРФЈ¬GPCRїЙ±»·ЦОЄЈµАаЈ¬·Ц±рКЗ:КУЧПємЦКСщјТЧеЎў·ЦГЪЛШКЬМеЎўґъР»РН№И°±ЛбКЬМеЎўр¤ёЅЛШКЬМеТФј°Frizzled/Taste2КЬМе[Јё]ЎЈ2012ДкЈ¬Еµ±ґ¶ы»ЇС§Ѕ±КЪУиБЛГА№ъїЖС§јТRobertєН BrianKЎЈТФ±нХГЛыГЗФЪGPCR№¦ДЬј°Ѕб№№СРѕїЦРµДН»іц№±ПЧЈ¬ЛыГЗµД№¤ЧчНЖ¶ЇБЛGPCRЅб№№ЙъОпС§µДґу·ўХ№Ј¬РВµДGPCRЅб№№І»¶П±»ЅвОц№«ІјЈ¬К№ИЛГЗ¶ФGPCRµДЅб№№ёґФУРФј°ЖдРЕєЕґ«µјµДёґФУРФУРБЛёьЙоИлµДИПК¶ЎЈ
ѕµдµДGPCRРЕєЕЧЄµјУЙЈЗµ°°ЧЅйµјЈ¬ЈЗµ°°ЧКЗУЙЈЗ¦БЎўЈЗ¦ВЎўЈЗ¦Г ИэёцСЗ»щЧйіЙµДТмФґИэѕЫМеЎЈЖдЦРЈ¬ЈЗ¦Б СЗ»щКЗѕЯУРGTPГё»оРФµД·ЦЧУїЄ№Шµ°°ЧЦК,ЈЗ¦В єН ЈЗ¦Г ФтТФ¶юѕЫМеРОКЅЅфГЬЅбєП,ЈЗ¦Б УРЈґЦЦСЗРН:ЈЗ¦БЈуЎўЈЗ¦БЈй/ ЈпЎўЈЗ¦БЈс/ Ј±Ј±єН ЈЗ¦Б12/13[Ј№]ЎЈГїЦЦСЗРНЅбєПІ»Н¬µДР§У¦Жчµ°°ЧЦКЈ¬Р§У¦Жчµ°°ЧЦКїЙТФЦ±ЅУ»тНЁ№эµЪ¶юРЕК№ЅшТ»ІЅј¤»оПВУОР§У¦(јыTable1).

ТФЈЗ¦БsОЄАэ:GPCRУлЕдМеЅбєПєу№№Пу·ўЙъёД±дЈ¬»о»ЇµДКЬМеУлЈЗµ°°ЧИэѕЫМе¦БСЗ»щЅбєПЈ¬µјЦВЈЗ¦БСЗ»щЙПµДGDPУл°ыЦКЦРµДGTPЅ»»»ЎЈЈЗ¦БСЗ»щУлКЬМеєНЈЗ¦В¦ГСЗ»щЅвАлЈ¬ЅбєПІўј¤»оПЩЬХЛб»·»ЇГё(ЈБЈГ)Ј¬Ѕш¶шК№Пё°ыДЪ»·БЧЛбПЩЬХ(cAMP)ЕЁ¶ИФцёЯЎЈcAMPУлµ°°ЧЦКј¤Гё ЈБ(PKA)µчЅЪСЗ»щЅбєПЈ¬µјЦВґЯ»ЇСЗ»щ±»КН·ЕІўИлєЛЎЈК№cAMPУ¦ґрФЄјюЅбєПµ°°ЧЦК(CREB)БЧЛб»ЇЈ¬БЧЛб»ЇµДCREBУлєЛДЪCREBЅбєПµ°°ЧЦК(CBP)МШТмЅбєПРОіЙёґєПОпЈ¬ёґєПОпУл°Р»щТтµчїШРтБРЅбєПЈ¬ј¤»о°Р»щТтЧЄВјЈ¬·ЦАлµДЈЗ¦В¦ГСЗ»щТІїЙТФј¤»оПВУОР§У¦·ЦЧУЎЈАэИзБЧЦ¬Гё ЈГ(PLC),PLCїЙЅ«БЧЦ¬хЈјЎґј-ЈґЈ¬Јµ-¶юБЧЛб (PIP2) Л®ЅвіЙµЪ¶юРЕК№јЎґјИэБЧЛб IP3)єН¶юхЈёКУН(DAG)ЎЄЎЄЎЄIP3ґМј¤Пё°ыДЪЦКНшКН·ЕCa2+ЅшИлПё°ыЦК»щЦКЈ¬К№°ыДЪCa2+ЕЁ¶ИЙэёЯЈ¬DAGј¤»оµ°°ЧЦКј¤ГёЈГ(PKC)Ј¬Ѕш¶шґҐ·ўMAPKРЕєЕНЁВ·(јыFig1)ЎЈ
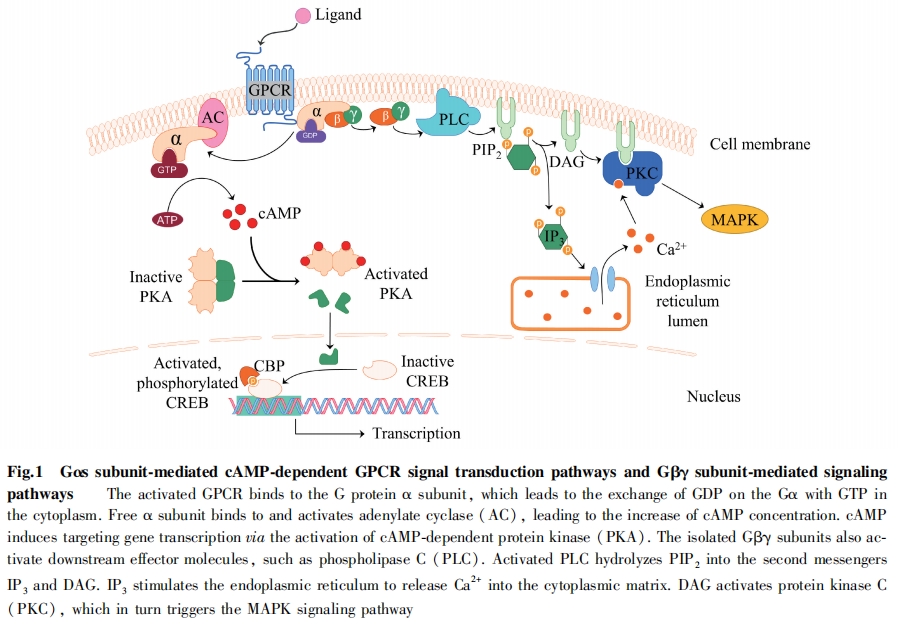
GPCRБнТ»ЦЦРЕєЕЧЄµј»ъЦЖУЙGPCRј¤Гё(GRKs) єНТЦЦЖµ°°Ч (arrestin) Ѕйµј[Ј№]Ј¬GPCRУлGRKsЅбєПµјЦВКЬМеБЧЛб»ЇЈ¬ґУ¶шЦХЦ№GPCRУлЈЗµ°°ЧµДП໥ЧчУГЈ¬Н¬К±Ј¬GPCRБЧЛб»Ї»№Жф¶ЇGPCRЈуУл ¦В-ТЦЦЖµ°°ЧЅбєПЎЈґ«Ні№ЫµгИПОЄЈ¬¦В-ТЦЦЖµ°°ЧКЗЧчОЄGPCRРЕєЕНЁВ·µДёє·ґАЎµчЅЪ»ъЦЖґжФЪµДЎЈЅьДкАґ·ўПЦЈ¬ЖдТІДЬЖф¶Ї·ЗЈЗµ°°ЧТААµµДПВУОРЕєЕНЁВ·[10-11]ЎЈДіР©ЕдМеУлGPCRЅбєПїЙµјЦВКЬМеСЎФсРФУлІ»Н¬СЗРНµДЈЗµ°°Ч»т¦В-ТЦЦЖµ°°ЧП໥ЧчУГЈ¬ґУ¶шУРЖ«ПтРФµШј¤»оДіР©МШ¶ЁµДРЕєЕНЁВ·Ј¬ХвР©ЕдМе±»іЖОЄЎ°Ж«ПтРФЕдМеЎ±[12]Ј¬ѕЯУРЖ«ПтРФµДЕдМеУлКЬМеЅбєПїЙј¤»о¦В-ТЦЦЖµ°°ЧНЁВ·Ј¬¦В-ТЦЦЖµ°°ЧїЙТФУлРн¶аПВУОР§У¦µ°°ЧЦКП໥ЧчУГЈ¬АэИзMAPKЎўPKBµИЈ¬ФЪGPCRРЕєЕЧЄµј№эіМЦР·ў»У№ШјьЧчУГ[13-14]Ј¬ХвТ»·ўПЦОЄ°РПтGPCRТ©ОпµДСР·ўМṩБЛРВµД·ЅПт:ѕЯУРЖ«ПтРФµДЕдМеДЬМШТмРФµШј¤»оЈЗµ°°Ч»тХЯ¦В-ТЦЦЖµ°°ЧЅйµјµДРЕєЕНЁВ·Ј¬Па±Иґ«НіЕдМеЖд±нПЦіцёьЗїµД°РПтРФЎЈХв¶ФМбёЯТ©Р§УРЦШТЄµДТвТеЎЈ
ЈІлДАаТ©Опј°ЖдСР·ўІЯВФ
¶алДЧчОЄТ©ОпѕЯУРРн¶а¶АМШµДУЕКЖ:ЈЁ1Ј©ґуБїМмИ»лДАаїЙЧчОЄЕдМеЧчУГУЪПё°ыД¤±нГжКЬМеЈ¬ѕЭґЛїЄ·ўµДлДАаТ©ОпЧчУГ»ъЦЖГчИ·ЎўЙъОп»оРФёЯЎўУГТ©јББїРЎЈ»(ЈІ)УлРЎ·ЦЧУТ©ОпПа±ИЈ¬лДАаТ©ОпІ»НЁ№эёОµДґъР»Ј¬ЖдґъР»ІъОпОЄ°±»щЛбЈ¬јёєхОЮ¶ѕРФЗТТ»°гІ»»бФЪМШ¶ЁЖч№ЩЧйЦЇЦРАЫ»эЈ¬ТтґЛё±ЧчУГ·ўЙъВКµНЈ¬°ІИ«РФєГЈ»(Јі)Улµ°°ЧЦКµИЙъОпґу·ЦЧУПа±ИЈ¬лДАаТ©ОпТЧУЪєПіЙЈ¬ИЭТЧУлФУЦК»тё±ІъЖ··ЦАлЈ¬ґї¶ИёЯЈ¬ЗТЖдСР·ўЦЬЖЪ¶МЈ¬ЙъІъіЙ±ѕµНЈ¬ТЧВъЧгЦРµИ№жДЈЦОБЖµДРиТЄ[15-17]ЎЈјшУЪХвР©БјєГµДТ©АнМШРФєН№МУРУЕКЖЈ¬лДАаТ©ОпіЙОЄЅьДкАґРВТ©ОпСР·ўµДИИµгЎЈДїЗ°Ј¬ТСУР50¶аЦЦ°РПтGPCRлДАаТ©Оп±»ГА№ъFDAЕъЧјЎЈЖдЦРґу¶аКэУГУЪґъР»РФјІІЎ»тЦЧБцµДЦОБЖЎѕ18ЎїЎЈ
лДАаТ©ОпµДСР·ўѕАъБЛИЛМелДїЄ·ўЎўМмИ»лДїЄ·ўєНЙъОпјјКхїЄ·ўЈіёцЅЧ¶ОЈ¬ЧоіхµД№ШЧўµгјЇЦРУЪ¶ФИЛМелДАај¤ЛШµДїЄ·ўєНАыУГЎЈДїЗ°Ј¬ЙПКРµДТ©Опґу¶а¶јКЗУГХвёцІЯВФєПіЙµДЈ¬ЧоОЄµдРНµДАэЧУКЗТИµєЛШ[16]Ј¬ЙПКАјН80-90ДкґъЈ¬СРѕїХЯАыУГЦШЧйDNAјјКхЦЖФміцёЯґї¶ИµДЙъОпєПіЙИЛТИµєЛШЈ¬ІўНЁ№э¶ФлДБґЅшРРРЮКОєПіЙБЛТИµєЛШАаЛЖОпЈ¬КµПЦБЛЛЩР§єНі¤Р§Ј¬ЙПКАјНД©Ј¬СРѕїИЛФ±Ѕ«МЅЛч·¶О§А©ґуµЅґжФЪУЪОўЙъОпЎўЦІОпЎў¶ЇОп¶ѕТєЦРµДМмИ»лДАа[19]ЎЈАэИзЈ¬УГУЪ2РНМЗДтІЎј°·КЕЦЦўЦОБЖµДExendin-4[19]Ј¬јМ¶ФМмИ»лДАаµДґу№жДЈНЪѕтЦ®єуЈ¬лДАаТ©ОпµДСР·ўЧЯЙПБЛАыУГ·ЦЧУЙъОпС§јјКхЅшРРЙёСЎєНїЄ·ўµД±ШУЙЦ®В·ЎЈ
АыУГ·ЦЧУЙъОпС§јјКхїЄ·ўлДАаТ©ОпµДБЅґу»щґЎКЗёЯ·б¶ИлДївµДЅЁБўєНєПККµДЙёСЎІЯВФЈ¬1985ДкЈ¬George P SmithНЁ№эЅ«лД¶ОИЪИлКЙѕъМе±нГжµ°°ЧЦКЈ¬їЄ·ўіцБЛКЙѕъМеХ№КѕјјКх[20]Ј¬1990ДкЈ¬George P SmithУлJamie K Scott Ѕ«Лж»ъРтБРлДХ№КѕФЪКЙѕъМе±нГжЈ¬ЅЁБўБЛКЙѕъМеХ№КѕЛж»ълДїв[21]ЎЈКЙѕъМеХ№КѕјјКхНЁ№э¶аВЦЗЧєНЙёСЎЈ¬їЙФЪёЯ·б¶ИЛж»ълДїв(·б¶ИїЙґп10µДѕЕґО·Ѕ)ЦРЙёСЎіцУл°Рµ°°ЧЦКѕЯУРёЯЗЧєНБ¦µДлД¶ОЈ¬НЁ№эDNAІвРтИ·¶ЁєтСЎлДµД±аВлРЕПўЈ¬ґУ¶шЅ«лД¶ОУл°Рµ°°ЧЦКµДЗЧєНБ¦КфРФУллД¶ОТЕґ«РЕПўПа№ШБЄЈ¬ЛжєуЈ¬РВµДmRNAХ№КѕјјКхЎўєЛМЗМеХ№КѕјјКхєНЅНДёХ№КѕјјКхµИІ»¶ПУїПЦЈ¬ЛьГЗУлКЙѕъМеХ№КѕјјКхТ»ЖрФЪ№эИҐ20ДкЦРІ»¶ПµГµЅ·ўХ№УлУЕ»ЇЈ¬АнВЫЙПДЬ№»Хл¶ФИОєО°Рµ°°ЧЦКЅшРР»щУЪЗЧєНБ¦µДёЯНЁБїЙёСЎ[22]Ј¬
2014ДкЈ¬ГА№ъScrippsСРѕїЛщLernerїОМвЧйАыУГТ»ЦЦІёИй¶ЇОпПё°ыПµЦРµДЧФ·ЦГЪРЕєЕПµНіЈ¬іЙ№¦ЙёСЎіцGLP-1RµДРВРНлДАај¤¶ЇјБP5ЧчОЄЦОБЖЈІРНМЗДтІЎµДЗ±ФЪТ©Оп[23]Ј¬ХвПојјКхНЁ№эСЄРЎ°еФґЙъі¤ТтЧУКЬМеїзД¤Ѕб№№Ут(PDGFR-TM)Ј¬Ѕ«єтСЎлД¶ОХ№КѕУЪІёИй¶ЇОпПё°ы±нГжЈ¬ЅбєПДї±кїзД¤µ°°ЧЦКµД№¦ДЬ±ЁёжПµНіЈ¬АыУГБчКЅПё°ыКхУлёЯНЁБїІвРтЙёСЎ°РПтїзД¤µ°°ЧЦКµДлДАај¤¶ЇјБ[23-25]Ј¬ёГПµНіНЁ№эФЪІёИй¶ЇОпПё°ыЦР№э±нґпДї±кїзД¤µ°°ЧЦКЈ¬¶ш·З№МПа»ЇµДЦШЧйµ°°ЧЦКЈ¬ЧоґуіМ¶ИЙП±ЈіЦБЛЖдМмИ»ЧґМ¬Ј¬ІўЗТАыУГ±ЁёжПµНіКµПЦБЛ»щУЪ№¦ДЬµДЙёСЎЎЈ
Јі °РПтЈЗµ°°ЧЕјБЄКЬМеµДлДАаТ©Опј°Жд·ўПЦ
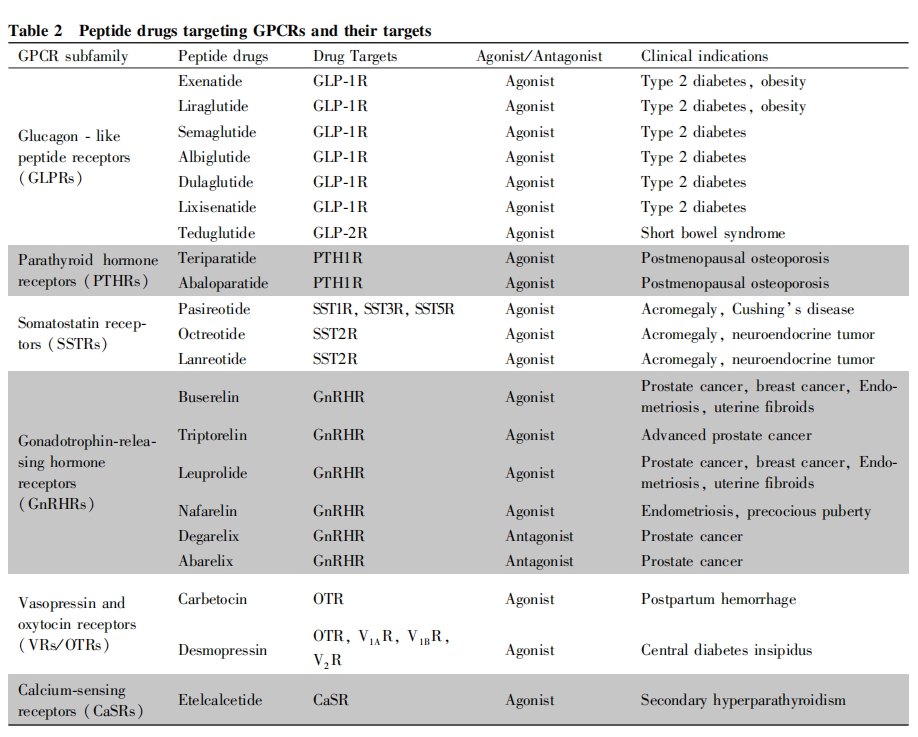
ИЛМеДЪУР¶аЦЦGPCRµДДЪФґРФ¶алДАаЕдМеЈ¬АэИзЅµёЖЛШЎўґЯІъЛШЎўЙъі¤ТЦЛШЎўТИёЯСЄМЗЛШСщлДЎўјУС№ЛШЎўјЧЧґЕФПЩЛШєНґЩРФПЩј¤ЛШКН·Еј¤ЛШµИЈ¬¶ФХвР©¶алДµДёДФмєНРЮКОКЗ¶алДТ©ОпїЄ·ўµДЦчТЄ·ЅПтЦ®Т»[18]ЎЈДїЗ°Ј¬ЙПКРµДЅ«Ѕь50ЦЦ°РПтGPCRµДлДАаТ©ОпµДЦчТЄ°Рµг°ьАЁТИёЯСЄМЗЛШСщлДКЬМеЎўјЧЧґЕФПЩЛШКЬМеЎўЙъі¤ТЦЛШКЬМеЎўґЩРФПЩј¤ЛШКН·Еј¤ЛШКЬМеЎўјУС№ЛШ/ ґЯІъЛШКЬМеєНёЖГфёРКЬМеµИЎЈУГУЪЦОБЖґъР»РФјІІЎЎўЙсѕПµНіјІІЎєН°©ЦўµИ¶аЦЦјІІЎЎЈТСЙПКРµД¶алДТ©Оп¶аКЗНЁ№э»ЇС§єПіЙ(АэИз№МПа¶алДєПіЙ·Ё)Ј¬»тХЯ»щТтЦШЧйјјКхєПіЙµДУµУРУлДЪФґРФ¶алДЕдМеПаЛЖРтБРµД¶алДАаЛЖОпј°ЖдСЬЙъОпЈ¬
ПВОДЅ«ЦШµгЅйЙЬјёАаСР·ўіЙ№¦ТСЙПКРµД°РПтGPCRлДАаТ©Оп(јыTable2)ЎЈ
3.1 °РПтТИёЯСЄМЗЛШСщлД1КЬМеµДлДАаТ©Оп
ТИёЯСЄМЗЛШСщлД-Ј±КЬМе(GLP-1R)ЦчТЄФЪТИПЩЦР±нґпЈ¬Ждј¤¶ЇјБАаТ©ОпКЗЅьДкАґЅµМЗТ©ОпСР·ўµДИИµгЈ¬GLP-1RµДМмИ»¶алДАаЕдМеТИёЯСЄМЗЛШСщлДЈ±( GLP-1) КЗ37ёц°±»щЛбµДлДЈ¬ЙгИлЖПМСМЗєуЈ¬і¦µАДЪ·ЦГЪЈМПё°ыєПіЙІў·ЦГЪGLP-1Ј¬ЧчУГУЪGLP-1RТэЖрПё°ыДЪcAMPФцјУЈ¬ґЩЅшЖПМСМЗУХµјµДТИµєЛШ·ЦГЪЈ¬ТЦЦЖТИёЯСЄМЗЛШµДКН·Е[26]Ј¬іэБЛЖдМШУРµДЖПМСМЗТААµµДТИПЩЧчУГНвЈ¬GLP-1»№·ў»УЧЕЅµµНКіУыєНґЩЅш±Ґё№ёРµДЦРКаЧчУГЈ¬Н»іцБЛЖдЧчОЄМЗДтІЎ»т·КЕЦЦўБЖ·ЁµДЗ±Б¦[27-28]Ј¬МмИ»GLP-1°лЛҐЖЪ¶МЈ¬»б±»МеДЪ¶юлД»щлДГёўф(DPP-IV) СёЛЩЅµЅвІў±»ЙцЗеіэЈ¬І»ККєПЦ±ЅУЧчОЄТ©ОпУ¦УГЈ¬ДїЗ°Ј¬ТСУР7ЦЦ¶алДАаGLP-1Rј¤¶ЇјБЙПКРЈ¬УГУЪЦОБЖ2РНМЗДтІЎЎЈ
3.1.1 Exendin-4ј°ЖдСЬЙъОп 1992ДкЈ¬EngµИґУGila¶ѕтбµД¶ѕТєЦР·ЦАліц Т» ЦЦМмИ»лД Exendin-4[29]ЎЈѕЯУРУлGLP-1ПаЛЖµДТ©АнС§МШРФЈ¬АэИзЈ¬їЙФцјУТИµєЛШ·ЦГЪЎўЅµµНСЄМЗЛ®ЖЅµИЈ¬УлGLP-1І»Н¬µДКЗЈ¬Exendin-4ТтЖдN-¶ЛµЪ2О»µД±ы°±ЛбМж»»ОЄёК°±ЛбЈ¬І»±»DPP-IVЅµЅвЈ¬ТтґЛѕЯУРёьі¤µД°лЛҐЖЪєНЅПЗїµДЙъОп»оРФ[30-31]Ј¬Exenatide(°¬ИыДЗлД)јґИЛ№¤єПіЙµД Exendin-4Ј¬°ьє¬39ёц°±»щЛбЈ¬ФЪ2005Дк»сµГFDAЕъЧјЈ¬КЗµЪЈ±ёцУГУЪЦОБЖЈІРНМЗДтІЎµДGPCR°РПтТ©[32]Ј¬ExenatideїЙТФУлGLP-1RЅбєПёД±дЖд№№ПуЈ¬ј¤»оAC,К№Пё°ыЦРcAMPФцјУЈ¬ґУ¶шґЩЅшЖПМСМЗТААµµДТИµєЛШ·ЦГЪЎўТЦЦЖТИёЯСЄМЗЛШ·ЦГЪєНФцјУТИµєЛШГфёРРФЈ¬ґпµЅЅµµНСЄМЗµДДїµД[31]ЎЈДїЗ°Ј¬Exenatide(УРГїИХ2ґО Exenatide BID) єНГїЦЬЈ±ґО(Exenatide QW)БЅЦЦјБРНЈ¬Exenatide BIDіэїЙУРР§ЅµµН»јХЯµДСЄМЗНвЈ¬»№ДЬ·ў»УЅµµНМеЦШµДЧчУГЈ¬МШ±рККєПЧчОЄєПІўПтРДРФ·КЕЦµДЈІРНМЗДтІЎ»јХЯµДЦОБЖТ©Оп[33]ЎЈµ«УЙУЪExenatide BIDРиТЄЖµ·±Ж¤ПВЧўЙдЈ¬їЙДЬµјЦВ»јХЯТАґУРФІо[18]Ј¬Exenatide QWКЗКЧёцКµПЦТ»ЦЬЈ±ґОёшТ©µДGLP-1Rј¤¶ЇјБЈ¬ЖдНЁ№эОўЗтјјКхЈ¬К№ExenatideФЪМеДЪ»єВэКН·ЕЈ¬·ў»Уі¤Р§ЅµМЗЧчУГЈ¬јМExenatide QWєуЈ¬ИЛГЗТІІЙУГБЛ¶аЦЦЖдЛыІЯВФАґМбёЯGLP-1Rј¤¶ЇјБµДСЄЅ¬ОИ¶ЁРФЈ¬ТФСУі¤Жд°лЛҐЖЪЎЈХвР©ІЯВФїЙґуЦВ·ЦОЄБЅЦЦ:Т»ЦЦКЗ¶ФExenatideµДРтБРЅшРРРЮКОЈ¬СУі¤ЖдСЄЅ¬°лЛҐЖЪЈ¬АэИзАыОчАлДЈ¬КЎВФБЛлДEx-enatideРтБРЦРµЪ38О»µДё¬°±ЛбІўФЪЖд ЈГ-¶ЛМнјУБЛЈ¶ёцАµ°±ЛбІР»щєНЈ±ёцхЈ°·»щЈ¬ХвР©РЮКОСУі¤БЛЖдСЄЅ¬°лЛҐЖЪ[34]
3.1.2 ТИёЯСЄМЗЛШСщлД1СЬЙъОп БнТ»ЦЦІЯВФФтІаЦШУЪРЮКОМмИ»µДGLP-1АэИзАыАВілДЎў°ў±ШВілДЎў¶ИАВілДєНЛчВнВілД,АыАВілДУЪ2010Дк»сµГFDAЕъЧјЈ¬ЖдЅ«МмИ»GLP-1ЙПµДµЪ34О»Аµ°±ЛбМж»»ОЄѕ«°±ЛбЈ¬ІўФЪµЪ26О»Аµ°±ЛбЙПФцјУБЛ1ёцБ¬ЅУ№И°±ЛбµДC16ЧШйµЦ¬·ѕЛбІаБґЈ¬ґУ¶шФцјУБЛЖдУлСЄЅ¬°Чµ°°ЧµДЅбєПЈ¬ЅµµНБЛDPP-IVµДГёЗРЧчУГєНЙцµДЗеіэЛЩВК[35]Ј¬іэТФУлExenatideАаЛЖµД»ъЦЖ·ў»УЅµМЗЧчУГНвЈ¬АыАВілДУлGLP-1RЅбєПєу»№ДЬСУ»єОёЕЕїХЈ¬ФЪЦОБЖ·КЕЦЦў·ЅГжР§№ыПФЦш[36]ЎЈ°ў±ШВілДєН¶ИАВілДѕщКЗТФИЪєПµ°°ЧЦКРОКЅЙъІъіцАґµДЎЈ°ў±ШВілДКЗНЁ№э»щТтЦШЧйјјКхАыУГЅНДёІъЙъµДЈ¬УлИЛСЄЗе°Чµ°°ЧИЪєПЈ¬ЖдЅ«МмИ»GLP-1ЙПµЪ2О»µД±ы°±ЛбМж»»ОЄёК°±ЛбТФµЦї№DPP-IVЅµЅвЈ¬¶ИАВілДКЗАыУГІёИй¶ЇОпПё°ыЕаСшІъЙъµДЈ¬№ІјЫБ¬ЅУµЅИЛIgG4-FcЦШБґЙПЈ¬ХвР©РЮКО·Ц±рЅµµНБЛХв2ЦЦТ©ОпЙцµДЗеіэВКЈ¬ФцјУБЛЖдТ©Ан»оРФµДіЦРшК±јд[37]ЎЈ
УлЙПКцјёЦЦGLP-1Rј¤¶ЇјБПа±ИЈ¬ЛчВнВілДЧоЦчТЄµДН»ЖЖКЗКµПЦБЛGLP-1Rј¤¶ЇјБµДі¤Р§ЦЖјБєНїЪ·юЦЖјБЈ¬2017ДкЈ¬ЛчВнВілДЧўЙдјБ±»FDAЕъЧјЙПКРЈ¬їЙСУі¤µЅГїЦЬЧўЙдЈ±ґОЈ¬Жді¤Р§»ъЦЖКЗ»щУЪ¶ФЅб№№µД»ЇС§РЮКОЈ¬ФЪЛчВнВілДЦРЈ¬МмИ»GLP-1µДµЪ8Ўў34О»°±»щЛб·Ц±р±»2-°±»щТм¶ЎЛбєНѕ«°±ЛбИЎґъЈ¬К№ЖдДЬУРР§±ЬГв±»DPP-IVЅµЅвЈ¬ІўЗТЈ¬ЖдЦРМмИ»GLP-1µДµЪ26О»Аµ°±Лб±»УІЦ¬ЛбхЈ»ЇЈ¬ДЬ№»К№ЖдУлИЛСЄЗе°Чµ°°ЧЅбєП,ґУ¶шСУі¤СЄЅ¬°лЛҐЖЪІў±ЬГв±»ЙцїмЛЩЗеіэЈ¬ХвР©РЮКО¶јСУі¤БЛ¶алДФЪМеДЪС»·µДК±јдЈ¬К№Жд°лЛҐЖЪїЙТФґпµЅ 7dЦ®ѕГ[38]ЎЈЛчВнВілДїЪ·юЦЖјБёХУЪ2019Дк±»FDAЕъЧјЙПКРЈ¬ЖдАыУГБЛEligen№«ЛѕСР·ўµД»щУЪґЩОьКХјБµДґу·ЦЧУµЭЛНјјКх:ґу·ЦЧУТ©Оп±»¶аёцґЩОьКХјБSNAC[8-(ЈІ-фЗ»щ±ЅјЧхЈ°·»щ) РБЛбДЖ]·ЦЧУ°ь№ьРОіЙЦ¬ЦКМеЅб№№Ј¬їЙ±Ј»¤¶алДТ©ОпІ»±»ОёЦРµДГёАаЅµЅв[38]
3.2 °РПтТИёЯСЄМЗЛШСщлД2КЬМеµДлДАаТ©Оп
ТИёЯСЄМЗЛШСщлД2КЬМе(GLP-2R)ЦчТЄ·ЦІјУЪОёі¦µАЧйЦЇЦРЈ¬НЁ№эcAMPТААµРФРЕєЕЧЄµјНѕѕ¶КµПЦ¶ФОёі¦µАµДµчїШЈ¬ФцЗїі¦µАУЄСшОьКХЎЈGLP-2RµДМмИ»лДАаЕдМеТИёЯСЄМЗЛШСщлДЈІ(GLP-2)КЗУЙ33ёц°±»щЛбЧйіЙµДі¦ФґРФ¶алДЈ¬УЙі¦µАДЪ·ЦГЪ LПё°ы·ЦГЪ,їЙ±»DPP-IVїмЛЩЅµЅвЈ¬°лЛҐЖЪ·ЗіЈ¶МЈ¬ЅцОЄ7min[39-40]ЎЈМж¶ИВілДКЗТ»ЦЦУЙDNAЦШЧйјјКхєПіЙµДGLP-2АаЛЖОпЈ¬2012Дк±»FDAЕъЧјОЄЦОБЖТААµі¦НвУЄСшЦ§іЦµДіЙИЛ¶Мі¦ЧЫєПХчµД№В¶щТ©[41]Ј¬¶Мі¦ЧЫєПХчЈЁSBSЈ©КЗТ»ЦЦє±јыµДѕЯУРЗ±ФЪЙъГьОЈПХµДОьКХІ»БјјІІЎЈ¬ЖдФТтКЗПИМмРФИ±ПЭЎўјІІЎµјЦВµДОьКХХП°»т№г·єКЦКхЗРіэµјЦВґуІї·Ці¦µА№¦ДЬЙҐК§[40-41]ЎЈµ±SBS»јХЯі¦µАОьКХУЄСшОпЦКЎўµзЅвЦКєНЛ®µДДЬБ¦І»ДЬВъЧг»ъМеРиТЄК±Ј¬РиТЄі¦НвУЄСшЦ§іЦЈ¬ФЪМж¶ИВілДЦРЈ¬МмИ»GLP-2µДµЪ2О»±ы°±Лб±»Мж»»ОЄёК°±ЛбТФµЦї№DPP-IVЅµЅвЎўСУі¤СЄЅ¬°лЛҐЖЪЈ¬ЖдїЙНЁ№эЧчУГУЪGLP-2RАґ·ў»УЙъОпС§ЧчУГЈ¬КµПЦ¶ФОёі¦µАµДµчїШЈ¬јхЙЩОёЕЕїХєН·ЦГЪЈ¬ІўґЩЅшРЎі¦ХіД¤ЙПЖ¤Пё°ыµДЙъі¤ЎўФцЦієНРЮёґЈ¬ґУ¶шФцјУРЎі¦ОьКХЎўјхЙЩё№Рє[40-42]ЎЈ
3.3 °РПтјЧЧґЕФПЩЛШ1КЬМеµДлДАаТ©Оп
јЧЧґЕФПЩЛШ1КЬМе( PTH1R)ЦчТЄ±нґпУЪЙцєН№ЗчАЈ¬ґжФЪ2ЦЦІ»Н¬µДёЯЗЧєНРФ№№Пу:ЈЗµ°°Ч·ЗТААµ№№Пу(R0)єНЈЗµ°°ЧТААµ№№Пу(RG)ЎЈУлR0№№ПуЗЧєНБ¦ёьёЯµДЕдМеЦчТЄј¤»о ¦В-ТЦЦЖµ°°ЧРЕєЕНЁВ·¶шТэ·ўКЬМеДЪНМЈ¬ґҐ·ўі¤К±іМРЕєЕ·ґУ¦Ј¬УлRG№№ПуЗЧєНБ¦ёьёЯµДЕдМеЦчТЄј¤»оGµ°°ЧЅйµјµДcAMPТААµРФРЕєЕЧЄµјНЁВ·Ј¬ґҐ·ўЛІК±РЕєЕ·ґУ¦[43]ЎЈјЧЧґЕФПЩј¤ЛШ(PTH) єНјЧЧґЕФПЩј¤ЛШПа№ШлД( PTHrP) КЗ№ЗґъР»№эіМЦРЦШТЄµДµчЅЪТтЧУЈ¬јЧЧґЕФПЩј¤ЛШПа№ШлДµДЈО-Д©¶Лє¬УРјЧЧґЕФПЩј¤ЛШН¬ФґРтБРЎЈ¶юХЯѕщїЙЧчУГУЪPTH1RНЁ№эcAMPТААµРФµДPKAРЕєЕЧЄµјНЁВ·µчЅЪ№ЗґъР»[44]ЎЈІ»Н¬µДКЗјЧЧґЕФПЩј¤ЛШЦчТЄµчЅЪёЖµДОИМ¬єН№ЗОьКХЈ¬¶шјЧЧґЕФПЩј¤ЛШПа№ШлДКЗґЩЅш№ЗРОіЙµД№ШјьлДЎЈ
3.3.1 јЧЧґЕФПЩј¤ЛШАаЛЖОп МШБўЕБлДКЗє¬УР34ёц°±»щЛбµДјЧЧґЕФПЩј¤ЛШАаЛЖОп,ТІКЗµЪ1ёц±»FDAЕъЧјЙПКРµДµчЅЪ№ЗґъР»µДлДАаТ©ОпЈ¬ЖдЦчТЄЧчУГУЪјЧЧґЕФПЩЛШ1КЬМеµДR0№№П󣬴ٽш ¦В-ТЦЦЖµ°°ЧµДЅбєП¶шµјЦВДЪНМ№эіМЈ¬їЙіЦРшµчЅЪcAMPє¬БїЈ¬ґУ¶шіЦРшРФј¤»оПВУОРЕєЕНЁВ·Ј¬Ѕйµјі¤К±іМРЕєЕЧЄµјЈ¬јУїм№ЗОьКХ[18Ј¬43-45]ЎЈ
3.3.2 јЧЧґЕФПЩј¤ЛШПа№ШлДАаЛЖОп °ў°НВелДКЗµЪ1ёц±»їЄ·ўµДјЧЧґЕФПЩј¤ЛШПа№ШлДАаЛЖОп,ѕЯУР34ёц°±»щЛбЎЈУЪ2017Дк±»FDAЕъЧјУГУЪѕшѕєуЕ®РФ№ЗЦККиЛЙЦўµДЦОБЖЎЈЖдЦчТЄЧчУГУЪјЧЧґЕФПЩЛШ1КЬМеµДRG№№ПуЈ¬Тэ·ўЛІК±µДcAMPє¬БїµДФцёЯЈ¬Ѕйµј¶МФЭµДРЕєЕ·ґУ¦Ј¬ґЩЅш№ЗРОіЙµДДЬБ¦і¬№э№ЗОьКХ[18,43]ЎЈХв ЈІ ЦЦТ©Оп°РПтН¬Т»КЬМејЧЧґЕФПЩЛШ1КЬМеЈ¬µ«УЙУЪЖд№№ПуЖ«єГРФґжФЪІоТмЈ¬ЛьГЗј¤»оН¬Т»°РµгµДПВУОРЕєЕНЁВ·І»Н¬Ј¬µјЦВ°ў°НВелД¶ФУЪѕшѕєуЕ®РФ№ЗЦККиЛЙЦўµДБЖР§УЕУЪ МШБўЕБлД[44]ЎЈХвТІЖфКѕФЪСР·ў°РПтGPCRµДлДАаТ©ОпК±У¦їјВЗЖд№№ПуЖ«єГРФЎЈ
3.4 °РПтЙъі¤ТЦЛШКЬМеµДлДАаТ©Оп
Йъі¤ТЦЛШКЬМе(SSTR) јТЧе°ьАЁSSTR1-5Ј¬№г·є·ЦІјУЪЦРКаЙсѕПµНіЎўґ№МеєНРн¶аНвЦЬЖч№Щ[46]Ј¬Йъі¤ТЦЛШКЬМеµДМмИ»ЕдМеЙъі¤ТЦЛШ(SST) УлКЬМеЅбєПїЙУХµјcAMPТААµРФРЕєЕЧЄµјНѕѕ¶Ј¬ТЦЦЖёчЦЦґЩЦЧБцЙъі¤µДј¤ЛШєНЙъі¤ТтЧУµДКН·ЕЈ¬ґУ¶шТЦЦЖ°©Пё°ыФцЦі»тУХµј°©Пё°ыµтНцЈ¬Йъі¤ТЦЛШ¶ФКЬМеѕЯУРєЬёЯµДЗЧєНБ¦Ј¬µ«ФЪСЄЅ¬ЦРµД°лЛҐЖЪ·ЗіЈ¶МЈ¬ЅцОЄ1-3min[47]ЎЈТ»ЦЦИЛ№¤єПіЙµДМмИ»Йъі¤ТЦЛШµД°ЛлДСЬЙъОпЎЄЎЄ»·ЧґЙъі¤ТЦЛШКЬМеј¤¶ЇјБ°ВЗълДїЙУГУЪЦОБЖЙсѕДЪ·ЦГЪЦЧБцєНЦ«¶Л·КґуЦў[46]ЎЈНЁ№эТэИлD-°±»щЛбЈ¬°ВЗълДСЄЅ¬°лЛҐЖЪїЙґп72-113min,ІўїЙСЎФсРФµШУлЙъі¤ТЦЛШКЬМе2єНЙъі¤ТЦЛШКЬМеЈµЅбєП[46]Ј¬°ВЗълДУлЙъі¤ТЦЛШКЬМеЅбєПєуЈ¬НЁ№эPLCЅйµјµДРЕєЕЧЄµјНЁВ·Ј¬ІъЙъµЪ¶юРЕК№IP3ІўїЙј¤»оLРНCa2+НЁµАЈ¬ТЦЦЖЙъі¤ј¤ЛШµДІъЙъЈ¬Pasireo-tide(ЕБИрлД)КЗБнТ»ЦЦЙъі¤ТЦЛШКЬМеј¤¶ЇјБЈ¬КЗГА№ъєНЕ·ГЛЕъЧјµДЦОБЖївРАЧЫєПХчµД№В¶щТ©[48].Pa-sireotide¶ФЙъі¤ТЦЛШКЬМеЈµµДЗЧєНБ¦ЅПёЯЎЈДЬТЦЦЖґЩЙцЙППЩЖ¤ЦКј¤ЛШ(ACTH)µД·ЦГЪЈ¬К№ївРАЧЫєПХч»јХЯµДЖ¤ЦКґј·ЦГЪјхЙЩ[49]ЎЈ
3.5 °РПтґЩРФПЩј¤ЛШКН·Еј¤ЛШКЬМеµДлДАаТ©Оп
ґЩРФПЩј¤ЛШКН·Еј¤ЛШКЬМе( GnRHR) ЦчТЄФЪґ№МеєНЙъЦіПµНіПа№ШµДЧйЦЇЖч№ЩЦР±нґпЈ¬ЖдМмИ»лДАаЕдМеґЩРФПЩј¤ЛШКН·Еј¤ЛШ (GnRH)КЗПВЗрДФ·ЦГЪІъЙъµДТ»ЦЦК®лДЙсѕј¤ЛШЈ¬ФЪЙъЦіµчїШЦР·ў»УЦШТЄЧчУГ[50]
3.5.1 ґЩРФПЩј¤ЛШКН·Еј¤ЛШКЬМеј¤¶ЇјБLeuprolide ґЩРФПЩј¤ЛШКН·Еј¤ЛШАаЛЖОпLeuprolide(ББ±ыИрБЦ)КЗґЩРФПЩј¤ЛШКН·Еј¤ЛШКЬМеј¤¶ЇјБЈ¬1985Дк±»FDAЕъЧјЙПКРЈ¬УГУЪЦОБЖј¤ЛШ·ґУ¦РФ°©ЦўЈ¬АэИзЗ°БРПЩ°©єНИйПЩ°©Ј¬ТІїЙУГУЪЦОБЖЖдЛьґЖј¤ЛШТААµРФјІІЎЈ¬АэИзЧУ№¬ДЪД¤ТмО»ЦўєНЧУ№¬јЎБц[51]ЎЈLeuprolideЦчТЄНЁ№эЈЗ¦БЈс/11УлGPCRПа№ШБЄЈ¬ј¤»оPLCЈ¬КН·ЕIP3єНDAGЎЈУХµјPKCј¤»оЈ¬ґЩК№ґ№МеЗ°Т¶КН·Е»ЖМеЙъіЙЛШ(LH)єНВСЕЭґМј¤ЛШ(FSH)Ј¬ЧоЦХНЁ№эПВЗрДФ-ґ№Ме-РФПЩЦбµДХэіЈЙъАнЧчУГЈ¬УХµјСЄЗеґЖ¶юґјєНШєНЄЛ®ЖЅµДТ»№эРФЙэёЯ[52]ЎЈ¶шПВЗрДФ-ґ№Ме-РФПЩЦбµДµчЅЪТААµУЪПВЗрДФґЩРФПЩј¤ЛШКН·Еј¤ЛШµДВціе·ЦГЪЈ¬µјЦВLeuprolideБ¬РшјёЦЬЦОБЖєуґЩРФПЩј¤ЛШКН·Еј¤ЛШКЬМеГфёРРФПВЅµ[53]Ј¬ ХвЦЦґЩРФПЩј¤ЛШКН·Еј¤ЛШКЬМе»оРФµДі¤ЖЪПВµчКЗLeuprolideЦОБЖµДЦчТЄ»ъЦЖЈ¬ЧоЦХµјЦВ»ЖМеЙъіЙЛШєНВСЕЭґМј¤ЛШ·ЦГЪјхЙЩЈ¬РФПЩ№¦ДЬјхНЛЈ¬ґУ¶шК№ґЖ¶юґјєНШєНЄЛ®ЖЅПФЦшЅµµНЈ¬ХвТ»µгУлЖдЛыGPCRµДј¤¶ЇјБІ»Н¬ЎЈ
3.5.2 ґЩРФПЩј¤ЛШКН·Еј¤ЛШКЬМеЮЧї№јБDegarelix ФЪј¤ЛШГфёРРНЗ°БРПЩ°©µДЦОБЖЦРЈ¬LeuprolideЛщµјЦВµДШєНЄЛ®ЖЅµДіхКјЙэёЯ·ґ¶ш»бК№З°БРПЩ°©µДЦўЧґјУЦШЈ¬ХвґЩК№СРѕїХЯЅшТ»ІЅїЄ·ўБЛґЩРФПЩј¤ЛШКН·Еј¤ЛШКЬМеЮЧї№јБЈ¬2008ДкЈ¬FDAЕъЧјБЛТ»ЦЦґЩРФПЩј¤ЛШКН·Еј¤ЛШКЬМеЮЧї№јБDegarelix(µШјУИрїЛ)Ј¬УГУЪЦОБЖНнЖЪЗ°БРПЩ°©[54]Ј¬DegarelixУлґЩРФПЩј¤ЛШКН·Еј¤ЛШКЬМеїЙДжРФЅбєПЈ¬ПВµчПё°ыДЪcAMPє¬БїЈ¬НЁ№эТЦЦЖcAMPТААµµДGPCRРЕєЕНЁВ·јхЙЩґЩРФПЩј¤ЛШµДКН·ЕЈ¬Ѕш¶шјхЙЩШєНЄµДКН·ЕТФЧиЦ№З°БРПЩ°©µДЙъі¤єН¶с»ЇЈ¬НЁ№эФЪлДБґµДµЪ5/6О»ТэИлЈР-ле»щ±Ѕ±ы°±ЛбФЪКµПЦТ©ОпЧчУГК±јдСУі¤µДН¬К±ТІ±ЬГвБЛТтЧй°·КН·ЕТэЖрµДі¬Гф·ґУ¦[55]Ј¬УРСРѕї±нГчЈ¬ФЪЦОБЖНнЖЪЗ°БРПЩ°©·ЅГжЈ¬DegarelixУЕУЪLeuprolideЎѕ56ЎїЎЈ
3.6 °РПтјУС№ЛШ/ґЯІъЛШКЬМеµДлДАаТ©Оп
јУС№ЛШКЬМе(VRs) °ьАЁV1ARЎўV1BRєНV2RЎЈV1ARЦчТЄФЪёОЎўСЄ№ЬЖЅ»¬јЎєНСЄРЎ°еЦР±нґпЈ¬ґЩЅшСЄ№ЬКХЛхєНСЄРЎ°еѕЫјЇЎЈV1BRЦчТЄФЪґ№МеЦР±нґпЈ¬їЙґЩЅшґЩЙцЙППЩЖ¤ЦКј¤ЛШµДКН·ЕЎЈV2RЦчТЄФЪЙцјЇєП№ЬЦР±нґпЎЈѕЯУРї№АыДтЧчУГ[57-59]ЎЈґЯІъЛШКЬМе(OTRs) ЦчТЄ·ЦІјУЪЧУ№¬єНИйПЩ[60]ЎЈјУС№ЛШєНґЯІъЛШѕщОЄѕЕлДЈ¬ЦчТЄНЁ№эЅбєПЖдКЬМеАґ·ў»УЧчУГЎЈV1ARЎўV1BRУлґЯІъЛШКЬМеЧчУГ»ъЦЖПаЛЖЈ¬ЦчТЄНЁ№эЕјБЄµДЈЗ¦БЈс/11ґМј¤PLCµД»оРФЈ¬КН·ЕIP3єНDAGЈ¬УХµјДЪЦКНшCa2+КН·Е[60,61]ЎЈ¶шV2RЦчТЄУлЈЗ¦БsЕјБЄЈ¬ј¤»оACІъЙъcAMPЈ¬УХµјPKAј¤»о[62]ЎЈїЁ±ґЛх№¬ЛШКЗИЛ№¤РЮКОµДґЯІъЛШАаЛЖОпЈ¬МмИ»ґЯІъЛШ·УфЗ»щЙПµДЗв±»јЧ»щИЎґъЈ¬°ллЧ°±ЛбІР»щЙПµД°±»щєНБт·Ц±р±»ЗвєНСЗјЧ»щИЎґъЈ¬ХвР©РЮКОСУі¤БЛїЁ±ґЛх№¬ЛШµДЧчУГК±јд[63]Ј¬їЁ±ґЛх№¬ЛШїЙУлЧУ№¬ЖЅ»¬јЎґЯІъЛШКЬМеЅбєПЈ¬УХµјЧУ№¬ЅЪВЙРФКХЛхЈ¬ФцјУЧУ№¬КХЛхЖµВКєНЗї¶ИЈ¬ККУГУЪїШЦЖ·ЦГдєуіцСЄ[63]Ј¬ИҐ°±јУС№ЛШКЗИЛ№¤єПіЙµДѕЕлД»ЇєПОпЈ¬2017Дк±»FDAЕъЧјУГУЪЦОБЖЦРКаРФДт±АЦўЈ¬Жд¶ФV2RЧчУГЧоЗїЈ¬їЙНЁ№эМбёЯЙцјЇєП№ЬЙПЖ¤Пё°ыcAMPЛ®ЖЅґЩК№ЙцСЄ№ЬКжХЕ·ў»Уї№АыДтЧчУГЈ¬ТтґЛУлМмИ»јУС№ЛШПа±ИЈ¬ИҐ°±јУС№ЛШї№АыДтЧчУГПФЦшФцЗїЈ¬¶ш¶ФЖЅ»¬јЎµДЧчУГИґјхИхЈ¬ґУ¶ш±ЬГвБЛёЯСЄС№ТэЖрµДІ»БјЧчУГ[64]ЎЈ

3.7 °РПтёЖГфёРКЬМеµДлДАаТ©Оп
ёЖГфёРКЬМе(CaSR)№г·є·ЦІјУЪјЧЧґЕФПЩЎўОёі¦µАЎўЙцЎў№ЗЧйЦЇµИІОУлµчЅЪМеДЪёЖОИМ¬µДЧйЦЇЖч№ЩЦРЈ¬ СЄёЖЛ®ЖЅЙэёЯј¤»оёЖГфёРКЬМеЈ¬ґУ¶шТЦЦЖБЛјЧЧґЕФПЩј¤ЛШµД·ЦГЪЎЈПа·ґЈ¬СЄёЖЛ®ЖЅµДЅµµН»бК№ёЖГфёРКЬМеµД»оРФКЬµЅТЦЦЖЈ¬ґЩЅшјЧЧґЕФПЩј¤ЛШµД·ЦГЪ[65].јМ·ўРФјЧЧґЕФПЩ№¦ДЬїєЅшЦў( SHPT) КЗВэРФЙцјІІЎ(CKD)іЈјыµДТ»ЦЦВэРФЅшРРРФІў·ўЦў[65]Ј¬SHPTµДМШХчКЗјЧЧґЕФПЩФцЙъТэЖрµДјЧЧґЕФПЩј¤ЛШЛ®ЖЅЙэёЯЈ¬ТФј°ёЖєНБЧµИїуОпЦКґъР»ТміЈ[65]Ј¬ХвР©їуОпЦКК§єв»бµјЦВСПЦШµДІў·ўЦўЈ¬АэИз№ЗчАјІІЎЎўИнЧйЦЇёЖ»ЇЎўСЄ№ЬёЖ»ЇєНРДСЄ№ЬІў·ўЦўµИ[66]Ј¬ О¬АїЁлДКЗёЖГфёРКЬМеµДТ»ЦЦРВРНі¤Р§лДј¤¶ЇјБЈ¬2017Дк±»FDAЕъЧјУГУЪЦОБЖіЙДкСЄТєНёОц»јХЯјМ·ўРФјЧЧґЕФПЩ№¦ДЬїєЅш[67]ЎЈ О¬АїЁлДЦ±ЅУЅбєПІўј¤»ојЧЧґЕФПЩЙПµДёЖГфёРКЬМеЈ¬НЁ№эPLCЅ«PIP2БСЅвіЙµЪ¶юРЕК№IP3єНDAGЈ¬ґУ¶шТЦЦЖјЧЧґЕФПЩЦРµДјЧЧґЕФПЩј¤ЛШ·ЦГЪЎЈ
Јґ ОКМвУлХ№Ны
лДАаТ©ОпТФЖдЙъОп»оРФёЯЎўµН¶ѕРФЎў°ІИ«РФєГµИМШµгіЙОЄБЛЅьДкТ©ОпСР·ўµДИИµгЈ¬УИЖдКЗЅб№№ЙПЅьєхОЮПЮµДїЙДЬРФК№Ц®ёьККєПУЪ°РПтGPCRЎЈИ»¶шЈ¬КЙѕъМеХ№КѕµИјјКхФЪєЬґуіМ¶ИЙПТААµУЪ»оРФЦШЧйµ°°ЧЦКµДЦЖ±ёУл№МПа»ЇЈ¬ДїЗ°ЙРІ»ДЬХл¶ФGPCRЎўАлЧУНЁµАЎў АТ°±Лбј¤ГёКЬМе(RTK)µИїзД¤µ°°ЧЦКЅшРРЙёСЎ[16,23]Ј¬БнНвЈ¬»щУЪЗЧєНБ¦µДЙёСЎ№эіМІўІ»ДЬЗш±рїзД¤µ°°ЧЦКµДј¤¶ЇјБєНЮЧї№јБЈ¬јґІ»ДЬЅшРР№¦ДЬЙёСЎ[23]ЎЈ
LernerїОМвЧйїЄ·ўµДЧФ·ЦГЪРЕєЕПµНіКЧґОКµПЦБЛІёИй¶ЇОпПё°ыЦРµДлДАаХ№Кѕј°№¦ДЬЙёСЎ,И»¶шПаЅПКЙѕъМеХ№КѕјјКх,ґЛПµНі»№І»ДЬКµПЦёЯ·б¶ИлДївµДЙёСЎЈ¬ФТтФЪУЪёГМеПµЦРлДїв·б¶ИКЗАґЧФє¬NNKјтІўГЬВлЧУµДPCRІъОпЈ¬Ждѕ№эЦКБЈ№№ЅЁЧЄ»ЇМбИЎЎўЦКБЈЧЄИѕЎўВэІЎ¶ѕїв°ьЧ°ЎўёРИѕІёИй¶ЇОпПё°ыµИТ»ПµБРІЩЧчєуЈ¬ЧоЦХХ№КѕУЪПё°ы±нГжµДЛж»ълДїв·б¶ИґжФЪґу·щПВЅµ(лДїв·б¶ИФЪТФЙПІЅЦиЦРІ»¶П¶ЄК§)Ј¬¶шлДїв·б¶ИёЯµНКЗЙёСЎУРР§єтСЎлДµД№ШјьТтЛШЈ¬ТтґЛЈ¬ФЪLernerИ·БўБЛІёИй¶ЇОпПё°ыПµлДАаХ№КѕЙёСЎјјКхЦ®єуЈ¬ДЬ·сФЪІёИй¶ЇОпПё°ыДЪЅЁБўёґФУлДївКЗµ±З°лДАаТ©ОпїЄ·ўµДТ»ґуМфХЅЎЈ
ОТГЗїОМвЧйїЄ·ўБЛТ»ЦЦ»щУЪПЯРФЛ«БґDNAµДЎ°УлГЕ(AND Gate)Ў±ВЯј»щТтПЯВ·ЙијЖРВІЯВФЈ¬НЁ№эPCR·ґУ¦Ѕ«НкХыµД»щТт±нґпДЈїй(Жф¶ЇЧУ-»щТт±аВлЗш-poly(ЈБ)ОІРЕєЕ)Ір·ЦОЄ2ёцПЯРФЛ«БґDNA·ЦЧУТэИлІёИй¶ЇОпПё°ыЈ¬ЖдФЪПё°ыДЪѕ·ЗН¬ФґД©¶ЛБ¬ЅУ(NHEJ),»тН¬ФґЦШЧй(HR)»ъЦЖБ¬ЅУФЪТ»ЖрЦШРВРОіЙНкХы»щТт±нґпДЈїйЈ¬КµПЦЎ°УлГЕЎ±ФЛЛгј°»щТт±нґп[68](јыFig.2)ЎЈУЙґЛЈ¬ОТГЗЅЁБўБЛТ»ЦЦФЪІёИй¶ЇОпПё°ыПµДЪТэИлёЯ·б¶ИлДївµД·Ѕ·ЁЈ¬ЛжЧЕ»щУЪІёИй¶ЇОпПё°ыПµµДёЯ·б¶ИлДїв№№ЅЁІЯВФµДїЄ·ўј°ЧФ·ЦГЪЙёСЎМеПµµДЅЁБўЈ¬°РПтGPCRµДлДАаТ©ОпЙёСЎМеПµЅ«І»¶ПНкЙЖІўЗчУЪіЙКмЈ¬ХвЅ«ОЄёь¶алДАаТ©ОпµДїЄ·ўЅЁБўјбКµµД»щґЎЎЈ
ГвФрЙщГчЈє±ѕОДОЄРРТµЅ»БчС§П°Ј¬°жИЁ№йФЧчХЯј°ФФУЦѕЛщУРЈ¬ИзУРЗЦИЁЈ¬їЙБЄПµЙѕіэЎЈОДХВ±кЧўУРЧчХЯј°ОДХВіцґ¦Ј¬ИзРиФД¶БФОДј°ІОїјОДПЧЈ¬їЙФД¶БФФУЦѕЎЈ
