±ΨΈΡΫΪ÷ΊΒψΧ÷¬έΙΐ»Ξ 10 ÷Ν 15 Ρξ÷–≥ωœ÷ΒΡΒ±¥ζΚœ≥…≤Ώ¬‘Θ§÷ΊΒψΙΊΉΔΕ‘Κœ≥…”–”ΟΒΡΖ¥”ΠΙΐ≥ΧΒΡΜζάμΖΫΟφΓΘ‘ΎΦρ“ΣΧαΦΑΚœ≥…ΒΡ“ΜΑψΖΫΖ®÷°ΚσΘ§Έ“Ο«ΫΪΗ≈ ωΈΣ¥ΌΫχκΡΜΖΜ·ΕχΧΫΥςΒΡΗς÷÷ΙΙœσΖΫΟφΓΘ’β–©ΖΫΟφΑϋά®ΫαΙΙ‘ΣΥΊ“‘ΦΑΆβ≤ΩΫπ τάκΉ”¥ΌΫχΦΝΓΘΈ“Ο«ΫΪΚ≠Η«”…Κ§ΝρΙΌΡήΆ≈ΫιΒΦΒΡΜΖΜ·ΓΔΜΖ ’Υθ≤Ώ¬‘ΓΔΒΰΒΣΜ·Έο - »≤ΧΰΜΖΦ”≥…ΓΔΜΖ±’ΚœΗ¥Ζ÷Ϋβ“‘ΦΑΕύΉιΖ÷ΜΖΜ·≤Ώ¬‘ΓΘΉνΚσΘ§Έ“Ο«ΫΪΫι…ήΈΣΚœ≥…ΜΖκΡΩβΕχ…ηΦΤΒΡ”Π”ΟΓΘΫπ τ¥ΏΜ·ΒΡΫΜ≤φ≈ΦΝΣ“‘ΦΑΚ§κΡΦϋ“‘ΆβΝ§Ϋ”ΒΡœΏ–‘«ΑΧεΒΡΜΖΜ·≤Μ‘Ύ±ΨΈΡΒΡΧ÷¬έΖΕΈßΡΎΓΘ
ΗυΨίΤδΙΌΡήΆ≈ΒΡ≤ΜΆ§Θ§κΡΩ…“‘Ά®ΙΐΥΡ÷÷≤ΜΆ§ΒΡΖΫ ΫΜΖΜ·ΘΚΆΖΈ≤ΜΖΜ·Θ®C ΕΥΒΫ N ΕΥΘ©ΓΔΆΖ≤ύΝ¥ΜΖΜ·ΓΔ≤ύΝ¥Έ≤ΜΖΜ·Μρ≤ύΝ¥≤ύΝ¥ΜΖΜ·Θ®ΆΦ 1aΘ©ΓΘ
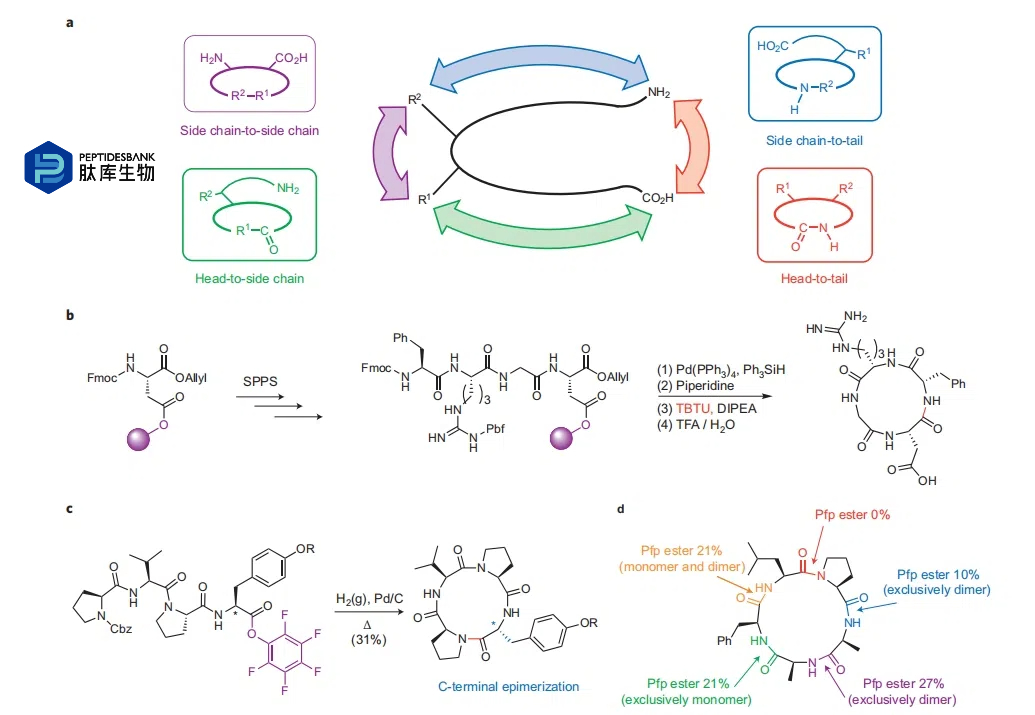
ΆΦ 1 | κΡ¥σΜΖΜ·ΒΡΆ®”ΟΚœ≥…ΩΦ¬«“ρΥΊΓΘaΘ§κΡ‘Ύ¥σΜΖ÷–Ω…Ρή ήΒΫœό÷ΤΒΡΥΡ÷÷ΖΫ ΫΓΘbΘ§¥”≤ύΝ¥”κΙΧœύ‘ΊΧεœύΝ§ΒΡΧλΕ§Α±ΥαΩΣ ΦΘ§≤…”ΟΨΏ”–»ΐΈ§’ΐΫΜ–‘ΒΡ±ΘΜΛΜυ≤Ώ¬‘Κœ≥…Κ§ RGD ΒΡΜΖΉ¥ΥΡκΡΒΡΙΧœύΚœ≥…ΓΘcΘ§ΥΡκΡ H-Pro-Val-Pro-Tyr-OPfp ‘ΎΜΖΜ·Ιΐ≥Χ÷– C ΕΥ≤νœρ“λΙΙΜ·ΒΡΈ ΧβΓΘCbz »Ξ±ΘΜΛ - ΜΖΜ·Ζ¥”Π‘Ύ 0.001 M ΒΡ≈®Ε»œ¬Ϋχ––Θ®CbzΘ§ή–―θτ ΜυΘ©ΓΘdΘ§ΜΖΉ¥ [Pro-Ala-Ala-Phe-Leu] Υυ”–Ω…ΡήΒΡΜΖΕœΝ―Θ§’Ι ΨΝΥΆ®Ιΐ Pfp θΞΜνΜ·Ϋχ––ΒΡΜΖΒΞΧεΜ·ΚΆ/ΜρΕΰΨέΜ·ΒΡΖ÷άκ≤ζ¬ ΓΘΜΖΜ·‘Ύ CHCl3/1 N aq.NaHCO3Θ®2ΘΚ1Θ©»ή“Κ÷–“‘ 0.0009 M ΒΡ≈®Ε»Ϋχ––ΓΘ
‘ΎΚœ≥…ΜΖκΡΒΡΗς÷÷ΖΫΖ®÷–Θ§Ήν÷’ΒΡΜΖΚœΖ¥”ΠΆ®≥Θ «ΡΎθΘΑΖΜ·ΓΔΡΎθΞΜ·Μρ–Έ≥…ΕΰΝρΦϋΓΘάΐ»γΘ§“Μ÷÷”––ßΒΡ≤ύΝ¥ΒΫ≤ύΝ¥ΒΡΚξΜΖΜ·Ζ¥”Π…φΦΑΧλΕ§Α±ΥαΜρΙ»Α±Υα≤ύΝ¥”κάΒΑ±Υα≤–Μυ÷°ΦδΒΡΥθΚœΖ¥”ΠΓΘΚξΜΖΜ·Ζ¥”ΠΉνΚΟ‘ΎΗΏœΓ ΆΕ»Θ®Ά®≥ΘΈΣ―«ΚΝΡΠΕϊ≈®Ε»Θ©œ¬Ϋχ––Θ§“‘ΨΓΝΩΦθ…Ό≤Μ±Ί“ΣΒΡΖ÷Ή”ΦδΙΐ≥ΧΘ§»γΒΆΨέΚΆΨέΚœΓΘ»ΜΕχΘ§’β–©Ζ¥”ΠΉΑ÷ΟΆυΆυœύΒ±Η¥‘”Θ§–η“Σ“ΜΗωΜρΕύΗωΉΔ…δ±ΟΓΘ
ΫΪΖ¥”Π–‘Ζ÷Ή”ΟΣΕ®”Ύ≤Μ»ή–‘ΨέΚœΈο…œΩ…≤ζ…ζ“Μ÷÷ΦΌœΓ Άœ÷œσΓΘœύΕ‘”Ύ»ή“Κ÷–ΡήΙΜΉ‘”…ά©…ΔΒΡΕάΝΔΖ÷Ή”Εχ―‘Θ§Ν§Ϋ”‘Ύ≤Μ»ή–‘ΨέΚœΈο…œΒΡΙΌΡήΆ≈±Υ¥Υœύ”ωΒΡΩ…Ρή–‘Ϋœ–ΓΓΘΙΧœύ¥σΜΖΜ·ΒΡ÷ς“Σ”≈ Τ‘Ύ”ΎΘ§Ά®≥Θ÷Μ–ηΦρΒΞΒΡœ¥Β”ΚΆΙΐ¬ΥΦ¥Ω…Άξ≥…¥ΩΜ·ΓΘ“ΣΫΪκΡ‘ΎΙΧœύ…œΜΖΚœΘ§Ήν≥Θ”ΟΒΡΖΫΖ® «Ά®ΙΐΧλΕ§Α±ΥαΜρΙ»Α±ΥαΒ»»ΐΙΌΡήΆ≈Α±ΜυΥαΒΡ≤ύΝ¥ΫΪœΏ–‘«ΑΧεΟΣΕ®‘ΎΙΧœύ…œΓΘ“ΣΙΙΫ®œΏ–‘κΡΓΔΆ―»Ξ N ΕΥΚΆ C ΕΥΒΡ±ΘΜΛΜυΆ≈ΓΔ“‘ΆΖΈ≤œύΝ§ΒΡΖΫ ΫΜΖΜ·Θ§ΉνΚσΫΪ≤ζΈο¥”ΙΧœύ÷ß≥÷Έο…œΝ―Ϋβœ¬ά¥Θ§÷Ν…Ό–η“Σ“ΜΗωΨΏ”–»ΐΗωΈ§Ε»’ΐΫΜ–‘ΒΡ±ΘΜΛΜυ≤Ώ¬‘ΓΘ
”–÷ζ”ΎΫΪΝΫΕΥΝ§Ϋ”‘Ύ“ΜΤπΒΡΙΙœσ“ΣΥΊ
¥σΜΖΜ·Ζ¥”ΠΒΡ≥…ΙΠ”κΖώ»ΓΨω”ΎœΏ–‘«ΑΧεΡήΖώ‘ΎΜΖΚœ«Α ΙΖ¥”Π–‘Ρ©ΕΥ‘ΎΩ’Φδ…œ‘Λœ»≈≈Ν–ΒΟΉψΙΜΫϋΓΘ’β÷÷‘Λœ»≈≈Ν–Μα–Έ≥…ΫœΗΏΒΡ”––ß≈®Ε»Θ§¥”ΕχΦθ…ΌΖ÷Ή”ΦδΖ¥”Π≤ζ…ζΒΡΗ±≤ζΈοΓΘ‘γ‘Ύ 1963 ΡξΘ§»ΥΟ«ΨΆ“―ΩΣ ΦΧΫΥςΖ¥”Π–‘Ρ©ΕΥΩ’ΦδΝΎΫϋ–‘ΒΡ÷Ί“Σ–‘Θ§Β± ±κΡΒΡΜΖΜ·«ψœρ”κΥυ≤βΒΟΒΡΫιΒγ≥Θ ΐ‘ωΝΩœύΙΊΝΣΓΘœΏ–‘«ΑΧεΒΡΖ÷Ή”ΙΙœσΨωΕ®ΝΥΥυ”– L –ΆΚΆΥυ”– D –ΆκΡΒΡΚξΙέΜΖΜ·Ζ¥”ΠΤΡΨΏΧτ’Ϋ–‘Θ§”…”Ύ«ψœρ”ΎΉν–ΓΜ·œ©±ϊΜυ’≈ΝΠΘ§ΥϋΟ«Ηϋ«ψœρ”Ύ≤…»Γ…λ’ΙΙΙœσΘ§¥”Εχ ΙΖ¥”Π–‘Ρ©ΕΥ±Υ¥ΥœύΨύΫœ‘ΕΓΘΕύΡξά¥Θ§»ΥΟ«ΩΣΖΔ≤ΔΉέ ωΝΥΕύ÷÷άϊ”ΟΙΙœσ‘ΛΉι÷·ά¥“ΐΒΦΚξΙέΜΖΜ·ΒΡ≤Ώ¬‘ΓΘ‘Ύ¥ΥΘ§’β–©≤Ώ¬‘±ΜΖ÷ΈΣΝΫάύΘΚΘ®iΘ©ΓΑΡΎ≤ΩΓ±ΙΙœσ“ΣΥΊΘ§Αϋά®Ε‘κΡΝ¥Ϋχ––Ι≤Φέ–ό Έ“‘¥ΌΫχΤδΡ©ΕΥΒΡΫαΚœΘΜΘ®iiΘ©ΓΑΆβ≤ΩΓ±ΙΙœσ“ΣΥΊΘ§…φΦΑ Ι”ΟΖ÷Ή”÷ßΦήΘ§’β–©÷ßΦήΦ»≤Μ”κκΡΙ≤ΦέΝ§Ϋ”Θ§“≤≤Μ‘ΎΜΖΚœΙΐ≥Χ÷–±ΜœϊΚΡΓΘΚσ’Ώ τ”ΎΡΘΑεΫιΒΦΒΡΨόΜΖΜ·Ζ¥”ΠΖΕ≥κΘ§’β «Μ·―ßΚœ≥…ΒΡ“ΜΗωΜν‘ΨΝλ”ρΓΘ
ΡΎ≤ΩΙΙœσ“ΣΥΊΓΘ‘ΎκΡΒΡΜΖΜ·Ιΐ≥Χ÷–Θ§Β±œΏ–‘«ΑΧεΒΡΗς÷÷ΫαΙΙ“ΣΥΊΡήΙΜ“‘Ήν–ΓΒΡ’≈ΝΠ¬ζΉψΙΐΕ…Χ§÷–ΝΫΕΥΒΡΫ«“Σ«σ ±Θ§ΜΖΜ·Ιΐ≥ΧΜα ήΒΫ«ύμυΓΘ¥ςΕύΡΎΚΆ ΖΟήΥΙ‘ΎΕ‘Έ¥’έΒΰΕύκΡΝ¥ΒΡΕΥΒΫΕΥΜΖ±’Ε·ΝΠ―ßΒΡ―–ΨΩ÷–ΒΟ≥ωΫα¬έΘ§Ϋœ≥ΛκΡΒΡΜΖ±’Ε·ΝΠ―ß”…κΡΡΎ«βΦϋΒΡ–Έ≥…ΚΆΥ≤ ±Π¬-’έΒΰΫαΙΙΨωΕ®Θ§’βΦ”ΥΌΝΥ–ρΝ–÷–œύΨύΫœ‘ΕΒΡ≤–Μυ÷°ΦδΫ”¥ΞΒΡΥ―ΥςΓΘΕ‘”ΎΚ§”–≥§Ιΐ 10 ΗωκΡΦϋΒΡΝ¥Θ§ΖΔœ÷ΜΖ±’ΥΌ¬ ≥Θ ΐ‘Ύ 20 ÷Ν 100 Ρ…ΟκΒΡ ±ΦδΖΕΈßΡΎ±μœ÷≥ωΟί¬…≥ΛΕ»“άάΒ–‘ΓΘΕ‘”ΎΫœ≥ΛΒΡκΡΘ§ΖΔœ÷κΡΡΎ«βΦϋΫΒΒΆΝΥΜΖ±’ΒΡΉ‘”…ΡήΓΘΫœΕΧκΡΒΡΜΖ±’Ε·ΝΠ―ß±δ¬ΐ“‘ΦΑ»±ΖΠκΡΡΎ«βΦϋΒΡΙέ≤λΫαΙϊΈΣΕΧΕύκΡΝ¥ΒΡΡΎ‘ΎΗ’–‘ΧαΙ©ΝΥ÷ΛΨίΓΘ
ΈΣΝΥΑο÷ζΫΪœΏ–‘κΡΒΡΝΫΕΥΝ§Ϋ”Τπά¥Θ§Μ·―ßΦ“Ο«Ή≈―έ”ΎΕΰΦΕΒΑΑΉ÷ ΫαΙΙΘ§”»Τδ «Ζ¥œρΉΣΫ«ΓΘ Βœ÷Ήν–ΓΕΥΒΫΕΥΨύάκΒΡ“Μ÷÷«…ΟνΖΫΖ® «‘ΎκΡΝ¥÷–Φδ“ΐ»κ“ΜΗωΥ≥ ΫθΘΑΖΦϋΘ§¥”Εχ–Έ≥…άύΥΤ”ΎΠ¬-ΉΣΫ«ΒΡΜυ–ρΓΘ”…”ΎΡήΙΜΆ§ ±»ίΡ…»ΐΦΕXaa-ProθΘΑΖΦϋΒΡΥ≥ ΫΚΆΖ¥ ΫΙΙœσΘ®Τδ÷–Xaa¥ζ±μ»ΈΚΈL-ΠΝ-Α±ΜυΥαΘ©Θ§Η§Α±Υα‘ΎΕύκΡ÷–ΒΡΖ¥œρΉΣΫ«÷–≥ωœ÷ΒΡ«ψœρΉνΗΏΓΘΨΓΙήΕΰΦΕΚΆ»ΐΦΕθΘΑΖΒΡ ΒΦ –ΐΉΣΡήάίΦΗΚθΟΜ”–≤ν±πΘ§ΒΪΒΣ‘≠Ή”…œΒΡΕνΆβ»Γ¥ζΜυœϊ≥ΐΝΥ“ΜΗωΙΙœσœύΕ‘”ΎΝμ“ΜΗωΙΙœσΒΡ¥σΝΩΡήΝΩΤΪ≤νΓΘ‘Ύ–μΕύΒΑΑΉ÷ ΨßΧεΫαΙΙ÷–ΕΦΙέ≤λΒΫΝΥΗ§Α±ΥαΒΡΥ≥ ΫθΘΑΖΦϋΓΘ‘Ύ“ΜœνΨ≠Βδ―–ΨΩ÷–Θ§¬όΧΊΦΑΤδΆ§ ¬άϊ”Ο’β“ΜΧΊ–‘ Βœ÷ΝΥ»ΐΗ§Α±ΥαΒΡΜΖΜ·ΓΘ
Κ§”–“λ–ΐΥΪΗ§Α±ΥαΒΞ‘ΣΒΡœΏ–‘κΡ «ΜΖΜ·Ζ¥”ΠΒΡΦΪΦ―ΒΉΈοΘ§’β «”…”Ύ d-Pro-l-Pro ΡΘΑεΨΏ”–Κή«ΩΒΡ”’ΒΦΠ¬-ΖΔΦ–ΫαΙΙΒΡΧΊ–‘ΓΘΫη÷ζ’β“ΜΤ§ΕΈΉιΉΑΒΡΜΖκΡΘ§¬ό±ω―ΖΦΑΤδΆ§ ¬ΡήΙΜΨΪ»Ζ÷Ίœ÷ΩΙΧεΤ§ΕΈΨßΧεΫαΙΙ÷–ΜΞ≤ΙΨωΕ®«χΜΖΒΡΒδ–ΆΙΙœσΓΘ
ΫΪΤδΥϊ d –ΆΑ±ΜυΥα≤τ»κ»Ϊ l –ΆκΡ÷–“≤“―÷ΣΜα≤ζ…ζ”’ΒΦΉΣΫ«ΒΡΉς”ΟΓΘ ΒΦ …œΘ§’β“Μ≤Ώ¬‘“―±Μ”Ο”ΎΧαΗΏΗς÷÷κΡΜΖΜ·ΒΡ≤ζ¬ ΓΘΕ‘”ΎΫω‘ΎΡ©ΕΥ≤–ΜυΒΡΠΝ-ΧΦΙΙ–Ά…œ≤ΜΆ§ΒΡΝΫΗωΖ«Ε‘”≥“λΙΙΧεΕΧκΡ–ρΝ–Θ§ΜΖΜ·Ηϋ”–άϊ”Ύ‘ΎΡ©ΕΥΆ§ ±Κ§”– d –ΆΚΆ l –Ά≤–ΜυΒΡΖ«Ε‘”≥“λΙΙΧεΓΘΫω”… l –Ά≤–ΜυΉι≥…ΒΡκΡΘ§»τ»±ΖΠΤδΥϊ”’ΒΦΉΣΫ«ΒΡΫαΙΙΘ§Ά®≥Θ“ΣΒ»ΒΫ C Ρ©ΕΥΒΡΠΝ-ΧΦ“λΙΙΜ·ΈΣ d –ΆΙΙ–Ά≤≈ΜαΜΖΜ·ΓΘd –ΆΑ±ΜυΥαΕ‘κΡ¥σΜΖΜ·ΒΡ”Αœλ“≤“―Ά®Ιΐάμ¬έΡΘ–ΆΫχ––ΝΥ―–ΨΩΓΘ
N-ΦΉΜυΑ±ΜυΥαΕ‘κΡΝ¥÷ςΝ¥ΒΡΝΔΧεΜ·―ß”Αœλ”κΗ§Α±ΥαάύΥΤΓΘΥϋΟ«”–Ω…Ρή‘ΎκΡ–ρΝ–÷–“ΐ»κΥ≥ ΫθΘΑΖΦϋΘ§≤Δ«“Ζ«≥Θ Κœ”’ΒΦΠ¬-ΉΣΫ«ΓΘΩ≠ΥΙά’ΦΑΤδΆ§ ¬“―≥δΖ÷÷ΛΟςΝΥΫΪΥϋΟ«ΉςΈΣ”’ΒΦΉΣΫ«ΒΡ‘ΣΥΊ“ΐ»κΜΖκΡ÷–ΒΡ«ιΩωΓΘ
Χ’ΕΌΚΆΒΥ‘ΎΚœ≥…Χλ»Μ≤ζΈοΫ«ΚΘΟύΥΊθΘΑΖ ±Φ«¬ΦΝΥ“ΜΗω“ΐ»Υ»κ ΛΒΡάΐΉ”Θ§ΗΟάΐΉ”ΥΒΟςΝΥœΏ–‘«ΑΧεΙΙœσΒΡ÷Ί“Σ–‘ΓΘ’β÷÷Ά®Ιΐ–ό ΈΒΡΜΖΉ¥ΤΏκΡΚ§”–ΝΫΗωΗ§Α±Υα≤–ΜυΓΔ“ΜΗωύγΏρΜΖΚΆ“ΜΗω΅fΏρΏχΜΖΓΘΥϋ‘Ύ…ζΈοΚœ≥…Ιΐ≥Χ÷–“‘ΝΫ÷÷≤ΜΡήœύΜΞΉΣΜ·ΒΡΗ§Α±ΥαθΘΑΖΙΙœσ“λΙΙΧεΒΡΜλΚœΈο–Έ Ϋ¥φ‘ΎΓΘΉς’ΏΖΔœ÷Θ§‘Ύ¥σΜΖΜ·÷°«ΑœΏ–‘«ΑΧε÷– «Ζώ¥φ‘Ύ”…Υ’Α±Υα―ή…ζΒΡ΅fΏρΏχΜΖΨωΕ®ΝΥ’βΝΫ÷÷ΙΙœσ“λΙΙΧεΒΡΕ·ΝΠ―ßΖ÷≤ΦΓΘ
Έ±Η§Α±Υα «¥”ΥΩΑ±ΥαΚΆΥ’Α±ΥαΘ®Θ®4SΘ©-―θ‘”ΜΖΕΓΆι-4-τ»ΥαΘ©“‘ΦΑΑκκΉΑ±ΥαΘ®Θ®4RΘ©-ύγΏρΆι-4-τ»ΥαΘ©―ή…ζΕχά¥ΒΡ–ό Έ‘”ΜΖΑ±ΜυΥαΓΘΥϋΟ«Ήν≥θ±Μ“ΐ»κΉςΈΣΫαΙΙΤΤΜΒ–‘ΙΙΫ®ΡΘΩιΘ§“‘Ζά÷ΙκΡ‘ΎΙΧœύΚœ≥…Ιΐ≥Χ÷–ΨέΦ·ΚΆΉ‘ΒόΚœΓΘΥϋΟ«Ω…Ά®ΙΐΥΩΑ±ΥαΓΔΥ’Α±ΥαΜρΑκκΉΑ±Υα”κ»©ΜρΆΣ‘ΎΥα¥ΏΜ·œ¬ΜΖΚœΕχ»ί“ΉΜώΒΟΓΘΒ±±Μ’ϊΚœΒΫκΡΝ¥÷– ±Θ§’β–©≤–Μυ÷ς“Σ”’ΒΦΤδ«ΑΒΡθΘΑΖΦϋ–Έ≥…Υ≥ ΫΙΙœσΘ§¥”Εχ–Έ≥… VI –ΆΠ¬-ΉΣΫ«ΫαΙΙΓΘΕύΗω―–ΨΩ–ΓΉι“―ΫΪ’β–©≤–Μυ”ΟΉςΚœ≥…ΕΧΒΡΓΔ ή‘Φ χΒΡΜΖκΡΒΡ«Ω¥σΉΣΫ«”’ΒΦ‘ΣΥΊΓΘΤδ÷–“ΜΗωœ‘÷χΒΡάΐΉ” « Mutter Κœ≥…ΜΖ»ΐκΡΜΖ-[Pro-Thr(ΠΖMe,Mepro)-Pro]Θ§ΤδΤπ Φ‘≠ΝœΈΣ H-Pro-Pro-Thr(ΠΖMe,Mepro)-OHΘ®ΆΦ 2aΘ©ΓΘΗΟκΡ±Μ÷ΛΟςΡή”κ PyBOPΘ®±Ϋ≤Δ»ΐΏρ-1-―θΜυ»ΐΏΝΩ©ΆιΜυΝΉΝυΖζΝΉΥα―ΈΘ©‘ΎΗΏ≤ζ¬ œ¬Υ≤ΦδΜΖΚœΘ§«“ΈόΒΆΨέΈοΫαΙΙΘ§≈®Ε»ΗΏ¥ο 0.1 MΓΘ
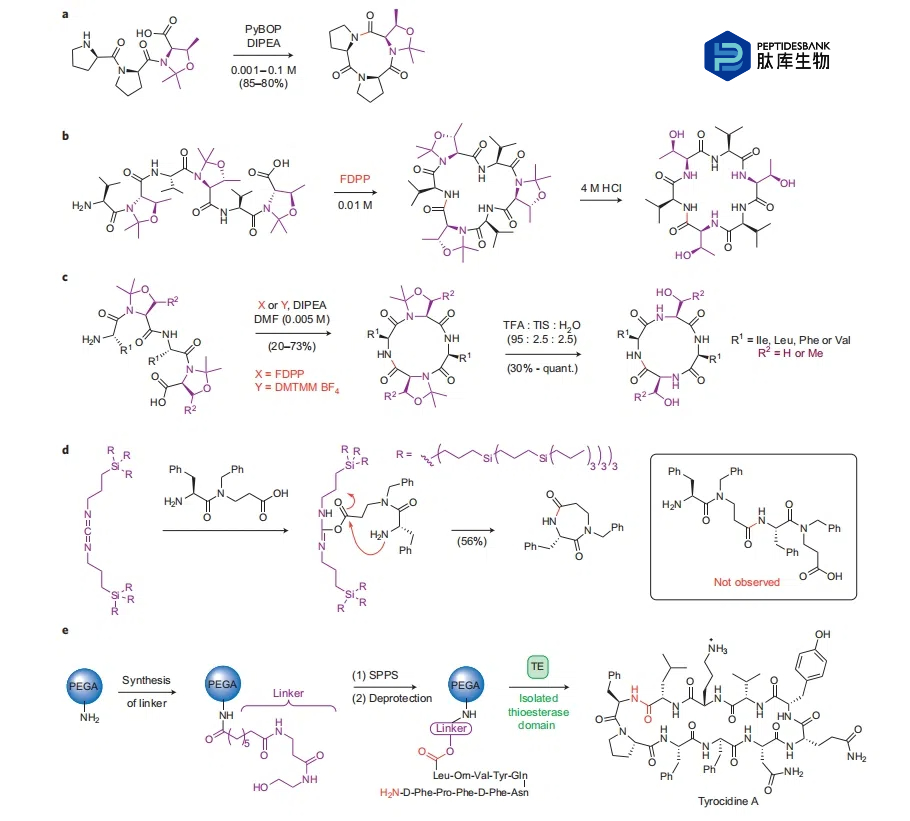
ΆΦ 2 | Ά®ΙΐΙΙœσΩΊ÷Τ¥ΌΫχκΡΜΖΜ·ΓΘaΘ§“Μ÷÷‘¥Ή‘ L-Υ’Α±ΥαΒΡΦΌΗ§Α±Υα–ό ΈΒΡ»ΐκΡΒΡ≈®Ε»ΈόΙΊΜΖΜ·ΓΘbΘ§≤ΜΚ§ΉΣΫ«”’ΒΦΦΝΒΡΜΖΦΚκΡΒΡΚœ≥…ΓΘ‘Ύ 0.01 M ΒΡ FDPP ≈®Ε»œ¬Θ§«Ε»κ»ΐΗωΦΌΗ§Α±Υα≤–ΜυΒΡœΏ–‘«ΑΧεΖΔ…ζΜΖΜ·ΓΘΜΖΜ·ΚσΘ§‘ΎΥ°–‘Υα–‘ΧθΦΰœ¬Θ§ΉΣΫ«”’ΒΦΒΡΦΌΗ§Α±Υα±ΜΜΙ‘≠ΈΣœύ”ΠΒΡΥ’Α±Υα≤–ΜυΓΘcΘ§Ά®Ιΐ FDPP Μρ DMTMM BF4 ≈ΦΝΣ ‘ΦΝ‘ΎΗΏœΓ ΆΕ»Θ®0.005 MΘ©œ¬ΜΖΜ·Θ§Κœ≥… C2 Ε‘≥ΤΒΡ»Ϊ L –ΆΜΖΜ·ΥΡκΡΘ§Τδ÷–ΑϋΚ§ΝΫΗωΫΜΧφΒΡΦΌΗ§Α±Υα≤–ΜυΓΘΜΖΜ·ΚσΘ§‘ΎΥα–‘ΧθΦΰœ¬Υ°ΫβΦΌΗ§Α±Υα“‘ ΆΖ≈œύ”ΠΒΡΧλ»ΜΑ±ΜυΥα≤–ΜυΓΘdΘ§–®–ΈΧΦΙηΆι ς÷ΠΉ¥ΧΦΕΰ―«ΑΖΆ®ΙΐΈΜΒψΗτάκΜζ÷ΤΜΖΜ·Ά§ΕΰΆΣΏΏύΚΓΘΈ¥Ιέ≤λΒΫœύ”ΠΒΡΕΰΨέΜ·≤ζΈοΓΘeΘ§Ζ÷άκΒΡΝρθΞΟΗΫαΙΙ”ρΆ®ΙΐΉΣθΘΜ·Κσ«Ω÷Τ‘ΛΜΖΜ·ΙΙœσ¥ΌΫχΩΙΨζΜΖκΡΒΡΖ¬…ζΚœ≥…ΓΘ“Μ÷÷ΙΧœύœΏ–‘«ΑΧεΓΘDIPEAΘ§N,N-Εΰ“λ±ϊΜυ““ΑΖΘΜDMTMM BF4Θ§4-(4Θ§6-ΕΰΦΉ―θΜυ-1Θ§3Θ§5-»ΐύΚ-2-Μυ)-4-ΦΉΜυ¬πΏχφfΥΡΖζ≈πΥα―ΈΘΜPEGAΘ§Ψέ““Εΰ¥ΦΨέ±ϊœ©θΘΑΖΙ≤ΨέΈοΘΜSPPSΘ§ΙΧœύκΡΚœ≥…ΘΜTFAΘ§»ΐΖζ““ΥαΘΜTISΘ§»ΐ“λ±ϊΜυΙηΆιΓΘ
Ι”ΟΈ±Η§Α±ΥαΉςΈΣΙΙœσΉΣΫ«”’ΒΦΦΝΒΡ“ΜΗωΕνΆβΚΟ¥Π «Θ§‘ΎΜΖΜ·÷°ΚσΘ§ΥϋΟ«Ω…“‘‘ΎΥα–‘ΧθΦΰœ¬±ΜΝ―ΫβΘ§¥”Εχ ΆΖ≈≥ωœύ”ΠΒΡΥΩΑ±ΥαΓΔΥ’Α±ΥαΜρΑκκΉΑ±Υα≤–ΜυΘ§¥”ΕχΒΟΒΫ≤ΜΚ§ΉΣΫ«”’ΒΦ‘ΣΥΊΒΡΜΖκΡΓΘJolliffe ΦΑΤδΆ§ ¬ΫΪ’β“Μ≤Ώ¬‘”Π”Ο”Ύ H-(Val-Thr)3-OH ΒΡΜΖΜ·Θ§’β «“Μ÷÷»±ΖΠΉΣΫ«”’ΒΦ‘ΣΥΊ«“ΈόΖ®Ά®Ιΐ≥ΘΙφΆΖΈ≤ΖΫ ΫΜΖΜ·ΒΡΝυκΡΘ®ΆΦ 2bΘ©ΓΘΉς’ΏΜΙΡήΙΜ÷ΛΟςΘ§Κ§”–ΝΫΗωΫΜΧφΈ±Η§Α±Υα≤–ΜυΒΡœΏ–‘ΥΡκΡΩ…“‘Ά®ΙΐΆΖΈ≤ΖΫ ΫΜΖΜ·Θ§¥”ΕχΒΟΒΫ ή‘Φ χΒΡ»Ϊ l –ΆΜΖΉ¥ΥΡκΡΘ®ΆΦ 2cΘ©ΓΘ”ΠΒ±÷Η≥ωΒΡ «Θ§ΨΓΙήΈ±Η§Α±ΥαΖ«≥Θ”–”ΟΘ§ΒΪΥϋΟ«ΒΡ“ΜΗω÷ς“Σ»±Βψ «”…”ΎΩ’Φδ”ΒΦΖΘ§NH Έ±Η§Α±ΥαΦΪΡ―θΘΜ·ΓΘΈΣΝΥΚœ≥…’β–© N-θΘΜ·‘”ΜΖΘ§ΉνΒδ–ΆΒΡΖΫΖ® « Ήœ»÷Τ±ΗΕΰκΡΤ§ΕΈΘ®Κ§ΥΩΑ±ΥαΜρΥ’Α±ΥαΘ©Θ§»ΜΚσ–Έ≥…Έ±Η§Α±ΥαΓΘ
Άβ≤ΩΙΙœσ“ΣΥΊΓΘ”Ο”ΎΗ®÷ζκΡ¥σΜΖΜ·ΒΡΆβ≤ΩΡΘΑεΜυ”ΎΈΜΒψΗτάκΜζ÷ΤΖΔΜ”Ής”ΟΓΘΨέΚœΈο÷ßΦήΡήΙΜ–Έ≥…Ϋω‘ –μ“ΜΧθœΏ–‘κΡΖ÷Ή”“Μ¥ΈΫχ»κ≤ΔΜΖΜ·ΒΡΖ¥”Π«ΜΓΘ’β÷÷ΕάΧΊΒΡΡ…ΟΉΜΖΨ≥ΫΪκΡ”κ÷ςΧε»ή“ΚΗτάκΩΣά¥ΓΘ“ρ¥ΥΘ§’β–©ΡΎ≤Ω«ΜΧεΥυ≤ζ…ζΒΡΗτάκ–ß”Πœ‘÷χΫΒΒΆΝΥΜΖΙ―ΨέΜ·ΒΡΩ…Ρή–‘ΓΘΖΕΓΛ¬μΕϊ»ϊΈΡΦΑΤδΆ§ ¬Ά®ΙΐΩΣΖΔΧΦΙηΆι ς÷ΠΉ¥ΧΦΕΰ―«ΑΖ”Π”ΟΝΥ’β“Μ≤Ώ¬‘ΓΘ’β–©–®–Έ≈ΦΝΣ ‘ΦΝΆ®ΙΐΤδ ς÷ΠΉ¥ΒΡΧεΜΐ≤ζ…ζΈΜΒψΗτάκ–ß”ΠΘ§Ά®ΙΐΤδ C ΕΥ≤ΕΜώΡΎθΘΑΖΜ·«ΑΧεΘ®ΆΦ 2dΘ©ΓΘΉς’Ώ÷ΛΟςΝΥ’β–© ς÷ΠΉ¥ΧΦΕΰ―«ΑΖΡήΙΜΜΖΜ·ΤΏ‘ΣΥΪΡΎθΘΑΖΘ®Ά§ΕΰΆΣΏΏύΚΘ©Θ§ΕχΆ®Ιΐ¥ΪΆ≥ΡΎθΘΑΖΜ·ΖΫΖ®ΚήΡ―ΜώΒΟ¥ΥάύΜ·ΚœΈοΓΘ
Χ©ΦΑΤδΆ§ ¬ΩΣΖΔΝΥΝμ“Μ÷÷”Ο”Ύ‘ω«ΩΥΡκΡΡ―“‘ Βœ÷ΒΡΜΖΜ·Ζ¥”ΠΒΡΆβ≤ΩΡΘΑεΓΘΥϊΟ«ΒΡΖΫΖ®Μυ”ΎΖ÷Ή””ΓΦΘΩ’«ΜΓΘ’β÷÷≤Ώ¬‘ «Ά®ΙΐΡ…ΟΉΦΕΩ’«ΜΈ§≥÷œΏ–‘κΡ÷–Υ≥ ΫθΘΑΖΙΙœσΒΡΉΣΫ«ΫαΙΙά¥ Βœ÷ΒΡΓΘ
Ή‘»ΜΫγ÷–ΒΡ–μΕύ¥σΜΖκΡ «Ά®ΙΐΖ«ΚΥΧ«Χε…ζΈοΚœ≥…≤ζ…ζΒΡΓΘ“―÷ΣΖ«ΚΥΧ«ΧεΚœ≥…ΟΗΆ®ΙΐΝρθΞΦϋΝ§Ϋ”ΜνΜ·ΒΡœΏ–‘÷–ΦδΧεΘ§ΤδΖΫ ΫάύΥΤ”ΎΙΧœύΚœ≥…ΓΘΈ÷Εϊ ≤ΦΑΤδΆ§ ¬Οη ωΝΥ“Μ÷÷ΖΫΖ®Θ§Τδ÷–Ζ÷άκ≥ωΒΡΝρθΞΟΗ¥ΌΫχΙΧΕ®‘ΎΚœ≥…ΙΧΧε÷ß≥÷Έο…œΒΡœΏ–‘κΡΒΡΜΖΜ·ΓΘΗΟΜζ÷Τ…φΦΑœρΜν–‘ΈΜΒψΥΩΑ±ΥαΒΡΉΣθΘΜ·Θ§ΥφΚσ‘ΎΖ÷Ή”ΡΎΑ±ΜυΡ©ΕΥ«ΉΚΥ ‘ΦΝΒΡΙΞΜςœ¬Ά―θΘΜ·ΓΘ’βœν―–ΨΩ±μΟςΘ§Ζ÷άκ≥ωΒΡΝρθΞΟΗΡήΙΜΫΪΙΧœύΝ§Ϋ”ΒΡΝρθΞΚΆθΞΝ§Ϋ”ΒΡœΏ–‘κΡ«Ω÷Τ≥…‘ΛΜΖΜ·ΙΙœσΓΘ’β÷÷Ά®”ΟΒΡΜ·―ßΟΗΖΫΖ®Ω…”Ο”Ύ…ζ≤ζ«Ω–ßΩΙΨζΦΝΘ®ΆΦ 2eΘ©ΓΘ–κΚΊΫΪ tRNA θΘΜ·ΚΥΟΗ”κ÷ΊΉιΈόœΗΑϊΖ≠“κœΒΆ≥ΒΡΫαΚœ Ι”ΟΘ§“―”Ο”Ύ÷Τ‘λΆ®ΙΐΝρΟ―ΦϋΝ§Ϋ”ΒΡκΡ¥σΜΖΓΘ
Ϋπ τάκΉ”Η®÷ζΜΖΜ·
Νμ“Μ÷÷Μυ”ΎΆβ≤ΩΖ«Ι≤ΦέΗ®÷ζΦΝ¥ΌΫχκΡ¥σΜΖΜ·ΒΡ≤Ώ¬‘…φΦΑΫπ τάκΉ”ΒΡ Ι”ΟΓΘ’β“Μ≤Ώ¬‘ΒΡΝιΗ–‘¥”Ύ“Μ–©Χλ»Μ¥φ‘ΎΒΡΜΖκΡΘ§»γΗΥΨζκΡΓΔΆΏάϊΡαΟΙΥΊΚΆΑΔΡ…ΥϊΡακΡ ««Ω–ßΒΡάκΉ”‘ΊΧεΘ§≤Δ«“‘ΎΧεΡΎΡή”κΫπ τάκΉ”–Έ≥…Έ»Ε®ΒΡΗ¥ΚœΈοΓΘ±¥ΩΥΦΑΤδΆ§ ¬ Ή¥Έ’Ι ΨΝΥάϊ”ΟΫπ τάκΉ” ΙκΡΙΙœσ‘ΛΉι÷·“‘¥ΌΫχ¥σΜΖΜ·ΒΡ ΒάΐΘ®ΆΦ3aΘ©ΓΘΥϊΟ«ΖΔœ÷Θ§‘ΎΦν–‘ΧθΦΰœ¬Θ§Ά®ΙΐΫπ τάκΉ”ΫιΒΦΒΡΝΫΗωΈ¥ΜνΜ·ΒΡΕΰκΡΦΉθΞΒΡΕΰΨέΜ·Θ§Ω…“‘ΙΙΫ®C2Ε‘≥ΤΒΡΜΖΥΡκΡΓΘ Βœ÷ΥΪΆΖΈ≤ΥθΚœΒΡ«ΑΧα «ΝΫΗωΕΰκΡθΞ±Ί–κ Ήœ»“‘Ζ¥ ΫΖΫ Ϋ≈δΈΜΒΫΫπ τ÷––ΡΘ®”κΥ≥ Ϋ≈δΚœΈο¥Π”ΎΤΫΚβΉ¥Χ§Θ©Θ§¥”Εχ‘ –μ≈δΈΜΒΡΑ±ΜυΕ‘ΝΎΫϋΒΡΕΰκΡθΞΫχ––«ΉΚΥΙΞΜςΓΘ’β÷÷ΜΖΕΰΨέΜ·ΖΫΖ®Κ≠Η«ΝΥ¥”12‘ΣΒΫ18‘ΣΒΡΕύ÷÷ΜΖ¥σ–ΓΓΘΜΖκΡΘ®”…ΠΝΚΆΠ¬Α±ΜυΥαΉι≥…Θ©Έό–ηΗΏœΓ ΆΓΔ±ΘΜΛΜρΜνΜ·ΜυΆ≈Θ§“≤≤Μ–η“Σ≈ΦΝΣ ‘ΦΝΓΘΆ®Ιΐ”κ Β±ΒΡ―τάκΉ”≥ΝΒμΘ§Ω…Ζ÷άκ≥ωΫπ τ≈δΈΜΒΡΕΰ“θάκΉ”Θ§«“Ϋπ τάκΉ”Ω…Ά®ΙΐΥα–‘ΦΉ¥ΦΫβΫΜΜΜΈΣ÷ Ή”ΓΘ
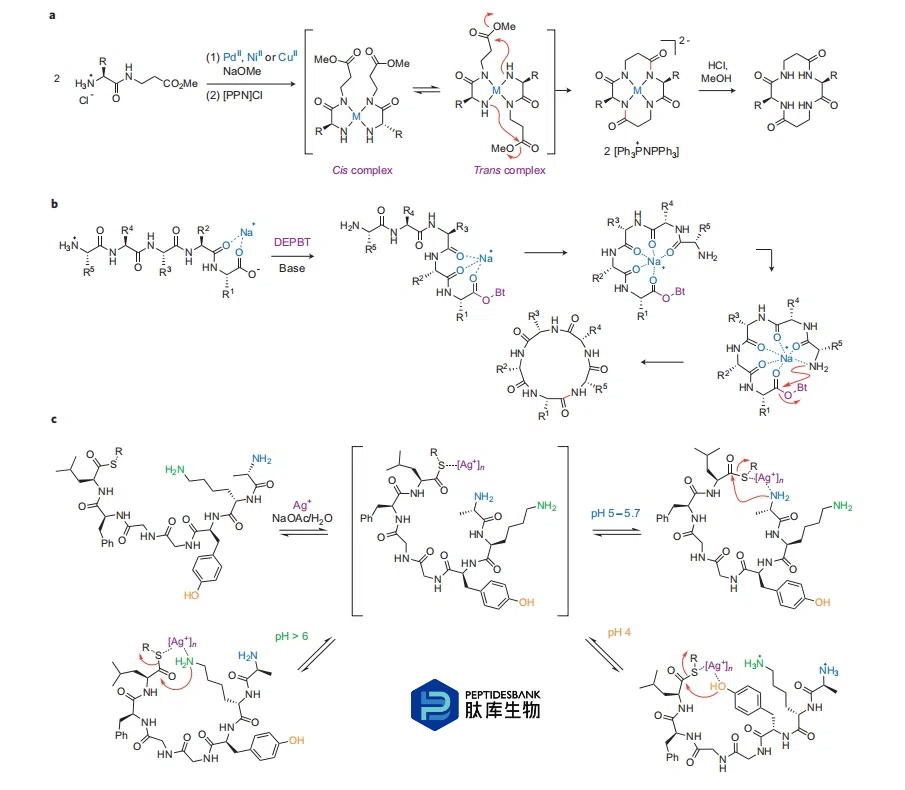 ΆΦ 3 | Ϋπ τάκΉ”‘ΎκΡ¥σΜΖΜ·÷–ΒΡ”Π”ΟΓΘaΘ§ΙΐΕ…Ϋπ τΫιΒΦΒΡΈ¥ΜνΜ·ΕΰκΡθΞΒΡΜΖΕΰΨέΜ·ΓΘbΘ§ΡΤάκΉ”Ά®ΙΐκΡΝ¥…œθΘΑΖ―θ‘≠Ή”ΒΡ¥°ΝΣ≈δΈΜά¥‘ω«ΩκΡΒΡΆΖΈ≤ΜΖΜ·ΓΘcΘ§Ag+άκΉ”ΫιΒΦΒΡκΡΝρθΞ¥σΜΖΜ·÷– pH “άάΒΒΡΜ·―ß―Γ‘ώ–‘ΓΘ‘Ύ pH 4 ±Θ§N ΕΥΚΆάΒΑ±Υα≤ύΝ¥ΒΡΑ±Μυ÷ Ή”Μ·Θ§”–άϊ”Ύ”κά“Α±Υα≤–ΜυΒΡΖ”―θ“θάκΉ”ΖΔ…ζ¥σΜΖΜ·ΓΘ‘Ύ pH 5 - 5.7 ÷°ΦδΙέ≤λΒΫΆΖΈ≤ΜΖΜ·’Φ”≈ ΤΓΘ‘Ύ pH 6 “‘…œΘ§άΒΑ±Υα≤–ΜυΒΡ Π≈-Α±Μυ”κκΡΝ¥ΖΔ…ζΡΎθΘΑΖΜ·±δΒΟœ‘÷χΓΘDEPBTΘ§3-(Εΰ““―θΜυΝΉθΘ―θΜυ)-1Θ§2Θ§3-±Ϋ≤Δ»ΐύΚ-4(3H)-ΆΣΘΜPPNΘΜΥΪ(»ΐ±ΫΜυλΔ)―«ΑΖφfΓΘ
ΆΦ 3 | Ϋπ τάκΉ”‘ΎκΡ¥σΜΖΜ·÷–ΒΡ”Π”ΟΓΘaΘ§ΙΐΕ…Ϋπ τΫιΒΦΒΡΈ¥ΜνΜ·ΕΰκΡθΞΒΡΜΖΕΰΨέΜ·ΓΘbΘ§ΡΤάκΉ”Ά®ΙΐκΡΝ¥…œθΘΑΖ―θ‘≠Ή”ΒΡ¥°ΝΣ≈δΈΜά¥‘ω«ΩκΡΒΡΆΖΈ≤ΜΖΜ·ΓΘcΘ§Ag+άκΉ”ΫιΒΦΒΡκΡΝρθΞ¥σΜΖΜ·÷– pH “άάΒΒΡΜ·―ß―Γ‘ώ–‘ΓΘ‘Ύ pH 4 ±Θ§N ΕΥΚΆάΒΑ±Υα≤ύΝ¥ΒΡΑ±Μυ÷ Ή”Μ·Θ§”–άϊ”Ύ”κά“Α±Υα≤–ΜυΒΡΖ”―θ“θάκΉ”ΖΔ…ζ¥σΜΖΜ·ΓΘ‘Ύ pH 5 - 5.7 ÷°ΦδΙέ≤λΒΫΆΖΈ≤ΜΖΜ·’Φ”≈ ΤΓΘ‘Ύ pH 6 “‘…œΘ§άΒΑ±Υα≤–ΜυΒΡ Π≈-Α±Μυ”κκΡΝ¥ΖΔ…ζΡΎθΘΑΖΜ·±δΒΟœ‘÷χΓΘDEPBTΘ§3-(Εΰ““―θΜυΝΉθΘ―θΜυ)-1Θ§2Θ§3-±Ϋ≤Δ»ΐύΚ-4(3H)-ΆΣΘΜPPNΘΜΥΪ(»ΐ±ΫΜυλΔ)―«ΑΖφfΓΘ
Εύ÷÷ΦνΫπ τ“―±Μ÷ΛΟςΡή¥ΌΫχκΡΒΡΜΖΜ·ΓΘάΐ»γΘ§ο°―ΈΉν‘γ±Μ±®ΒάΡήΧαΗΏκΡ‘Ύ”–Μζ»ήΦΝ÷–ΒΡ»ήΫβΕ»Θ§¬ό±»ΦΑΤδΆ§ ¬ΖΔœ÷ΤδΡήΫιΒΦΦΗ÷÷ N-¬»““θΘΜ·ΓΔC-ΑκκΉΑ±ΥακΡΒΡ―Γ‘ώ–‘ΜΖΜ·Θ§’β–©κΡΡΘΡβΝΥ HIV-1 ΑϋΡΛΧ«ΒΑΑΉ gp120 ÷–ΒΡ C4 ΫαΙΙ”ρΓΘ‘ΎΥ°–‘ΧθΦΰœ¬Θ§’β–©κΡΫω“‘ΆΖΈ≤œύΝ§ΒΡΖΫ ΫΨέΚœΓΘ»ΜΕχΘ§‘Ύ LiCl/ΕΰΦΉΜυΦΉθΘΑΖ»ήΦΝΜλΚœΈο÷–Θ§Ά®Ιΐ C Ρ©ΕΥΑκκΉΑ±ΥαΝρ¥ΦΕ‘ N Ρ©ΕΥ¬»““θΘΜυΆ≈ΒΡ«ΉΚΥ»Γ¥ζΘ§Ιέ≤λΒΫΝΥΉ®“ΜΒΡΒΞΧεΜΖΜ·ΓΘ“ΕΦΑΤδΆ§ ¬±μΟςΘ§ΡΤάκΉ” Κœ¥ΌΫχœΏ–‘ΈεκΡΒΡΜΖΜ·Θ§ΕχΫœ¥σΒΡοΛάκΉ”Ρή”––ߥΌΫχΤΏκΡΒΡΜΖΜ·ΓΘΜυ”ΎΖ÷Ή”ΝΠ―ßΦΤΥψΘ§“ΕΦΑΤδΚœ÷χ’ΏΧα≥ωΘ§’β–©ΦνΫπ τάκΉ”Ά®Ιΐ Ήœ»”κκΡ C Ρ©ΕΥΒΡτ ΜυΚΆθΘΑΖΜυΆ≈ΫαΚœά¥¥ΌΫχκΡΒΡΚξΜΖΜ·Θ§Ά®ΙΐθΘΑΖ―θΒΡΫχ“Μ≤Ϋ≈δΈΜΓΘΝ¥…œΒΡ’β–©άκΉ”Μα¥Ό ΙœΏ–‘κΡ–Έ≥…“ΜΗω«ΩΒΡΉΣΫ«ΫαΙΙΘ§¥”Εχ Ι N ΕΥΚΆ C ΕΥΩΩΫϋΘ§±ψ”ΎΜΖΜ·Θ®ΆΦ 3bΘ©ΓΘ
ΧΖΚΆ’≈ΜΙάϊ”ΟΝΥΫπ τάκΉ”¥ΌΫχκΡ¥σΜΖΜ·ΒΡΡήΝΠΓΘ ή“χΘ®ΔώΘ©Ε‘ΝρΓΔΒΣΓΔ―θ«ΉΚΆΝΠΥ≥–ρΈΣ SΘΨNΘΨO ΒΡΤτΖΔΘ§“‘ΦΑ≤Φά≥ΩΥΚΆάνΙΊ”ΎΝρθΞλ ΜνΜ· Βœ÷”––ßΤ§ΕΈ≈ΦΝΣΒΡΩΣ¥¥–‘ΙΛΉςΘ§ΧΖΚΆ’≈ΡήΙΜ÷ΛΟςΘ§‘ΎΥ°ΜΚ≥ε»ή“Κ÷–Θ§Β±“χάκΉ”ΒΡΒ±ΝΩ ΐΈΣ»ΐΗωΜρΗϋΕύ ±Θ§œΏ–‘κΡΝρθΞΡήΙΜ«α“ΉΒΊΖΔ…ζΜΖΜ·ΓΘ‘Ύ¥Υ”Π”Ο÷–Θ§“χάκΉ”Ά§ ±ΤπΒΫλΊΜνΜ·ΦΝΚΆλ ΜνΜ·ΦΝΒΡΉς”ΟΓΘΆ®Ιΐ”κ C ΕΥΝρθΞΚΆ N ΕΥΑΖ≈δΈΜΘ§“χάκΉ”ΫΪΖ¥”Π–‘ΙΌΡήΆ≈≤ΕΜώ‘ΎΫϋΝΎΈΜ÷ΟΘ§¥”Εχ Βœ÷θΘΜυΉΣ“ΤΜΖΚœΓΘ“χάκΉ”ΒΡ«ΉΝρ–‘‘Ύ”κΝρθΞ¬γΚœ ±≤ζ…ζλ ΜνΜ·Θ§ ΙΤδ≥…ΈΣΗϋΚΟΒΡάκ»ΞΜυΆ≈Θ§¥”Εχ¥ΌΫχθΘΜυΉΣ“ΤΖ¥”ΠΓΘΈ¥±ΘΜΛΒΡκΡΩ…“‘‘Ύ»ή“Κ÷–ΒΞΕάΜρΉςΈΣκΡΜλΚœΈοΫχ––ΜΖΜ·ΓΘΒ±ΫΪά“Α±ΥαΚΆάΒΑ±ΥαΘ®’βΝΫ÷÷ «ΨΚ’υ–‘«ΉΚΥ≤–ΜυΘ©“ΐ»κκΡΒΡ÷ςΝ¥ ±Θ§¥σΜΖΜ·ΒΡΜ·―ß―Γ‘ώ–‘ΫΪ»ΓΨω”Ύ»ή“ΚΒΡ pH ÷ΒΓΘ»ΜΕχΘ§”κ»ΈΚΈΆ®ΙΐΗΏΕ»ΜνΜ·ΒΡ C ΕΥθΞΫχ––¥σΜΖΜ·ΒΡΖΫΖ®“Μ―υΘ§“χάκΉ”¥ΌΫχΜνΜ· ±Ω…ΡήΜα≥ωœ÷≤νœρ“λΙΙΜ·ΒΡΈ ΧβΘ§Εχ«“»γΙϊΜΖΚœΖ¥”ΠΜΚ¬ΐΫχ––Θ§’β“ΜΈ ΧβΜαΗϋΦ”―œ÷ΊΘ®ΆΦ 3cΘ©ΓΘ
ΝρΫιΒΦΒΡΜΖΜ·Ζ¥”Π
–μΕύΙΊ”ΎκΡ¥σΜΖΜ·ΒΡΚœ≥…―–ΨΩΕΦ¥”Ή‘»ΜΫγ÷–Φ≥»ΓΝιΗ–ΓΘΥφΉ≈”Ο”ΎκΡ≈ΦΝΣΒΡœ»Ϋχ ‘ΦΝΒΡ≤ΜΕœΖΔ’ΙΘ§’β–©Ζ¬…ζΖΫΖ®≤…”Ο C ΕΥτ ΜυΒΡΝρθΞΜνΜ·ΓΘ
ήΏδΏρ‘ΎΜνΜ·θΘΜυΒΡΥ°ΫβΚΆΉΣ“Τ¥ΏΜ·÷–ΥυΤπΒΡΤ’±ιΉς”ΟΒΡΤτΖΔΘ§ΜτΕΌΦΑΤδΆ§ ¬ΩΣΖΔΝΥ“Μ÷÷‘ΎΏδΏρ¥φ‘Ύœ¬Ά®ΙΐκΡΝρθΞΒΡ÷±Ϋ”Α±Ϋβά¥ Βœ÷ΜΖκΡΆΖΈ≤Κœ≥…ΒΡΖΫΖ®Θ®ΆΦ 4aΘ©ΓΘΏδΏρΒΡΉς”Ο±Μ»œΈΣ ««ΉΚΥ¥ΏΜ·ΦΝΘ§ΥϋΙΞΜςΝρθΞΒΡτ Μυ–Έ≥…Μν–‘θΘΜυΏδΏρ÷–ΦδΧεΘ§ΥφΚσ±ΜΝμ“ΜΗω«ΉΚΥ ‘ΦΝ≤ΕΜώΓΘ¥” 5 ΒΫ 11 Ηω≤–Μυ≥ΛΕ»ΒΡΗς÷÷κΡ±ΜΜΖΜ·Θ§ΖΔœ÷ΤδΥΌ¬ »ΓΨω”ΎΜΖΒΡ¥σ–ΓΓΘΆ®ΙΐΏδΏρ¥ΏΜ·ΒΡ C ΕΥΝρθΞ”κΥΩΑ±Υα≤–Μυ≤ύΝ¥τ«Μυ÷°ΦδΒΡ¥σΜΖΡΎθΞΜ·Ζ¥”ΠΘ§“‘ΝΦΚΟΒΡ≤ζ¬ Κœ≥…ΝΥΩ®Ιΰά≠κΡ B ΦΑΤδάύΥΤΈοΓΘ
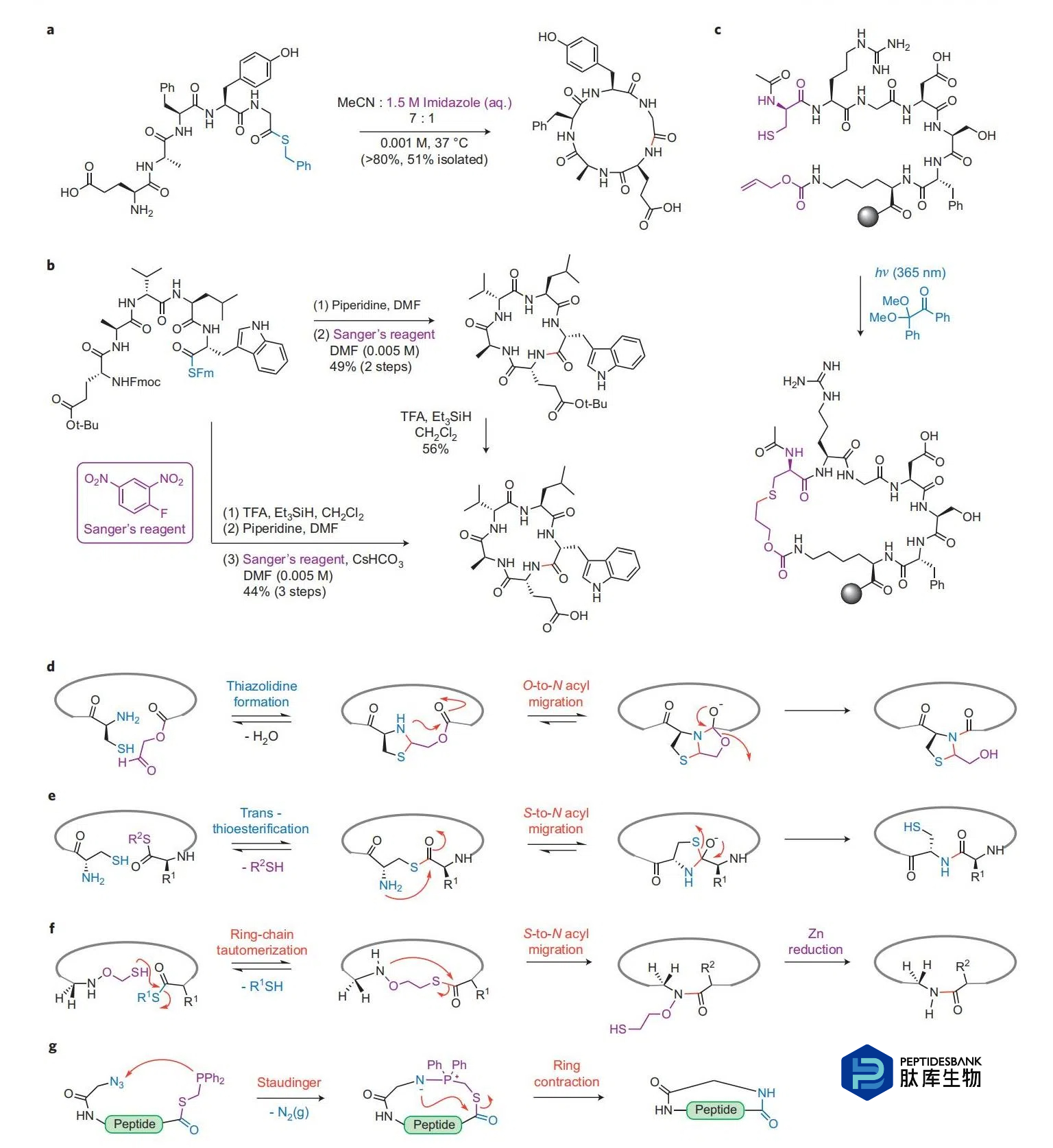
ΆΦ 4 | Ά®ΙΐΚ§ΝρΗ®÷ζΦΝΫιΒΦΒΡκΡ¥σΜΖΜ·ΓΘaΘ§”…ΏδΏρ¥ΏΜ·ΒΡΈεκΡΝρθΞΒΡΆΖΈ≤¥σΜΖΜ·ΓΘbΘ§ Ι”Ο Sanger ‘ΦΝΒΡΈεκΡΝρθΞΒΡΆΖΈ≤¥σΜΖΜ·ΓΘΗΟΜΖΜ·Ζ¥”Π”κΈ¥±ΘΜΛΒΡΙ»Α±ΥαΚΆ…ΪΑ±Υα≤ύΝ¥≤–Μυœύ»ίΓΘcΘ§‘ΎΙΧœύ…œΆ®ΙΐΝρœ©Ζ¥”ΠΫχ––ΒΡ≤ύΝ¥ΒΫΈ≤≤ΩΒΡ¥σΜΖΜ·ΓΘ’βΜΖΜ·Ζ¥”Π”… I –ΆΙβ“ΐΖΔΦΝ DMPAΘ®2,2-ΕΰΦΉ―θΜυ-2-±ΫΜυ±Ϋ““ΆΣΘ©‘Ύ 365 Ρ…ΟΉΙβ’’…δœ¬“ΐΖΔΓΘdΘ§Ά®Ιΐ–Έ≥…ύγΏρΏχΜΖΘ§ΥφΚσΖΔ…ζ O ΒΫ N ΒΡθΘΜυΉΣ“ΤΘ§N ΕΥΑκκΉΑ±Υα”κ C ΕΥΗ ”ΆθΞκΡΫχ––ΆΖΈ≤ΜΖΜ·ΓΘeΘ§‘≠ΈΜΜ·―ßΝ§Ϋ””Π”Ο”ΎκΡΒΡΆΖΈ≤ΜΖΜ·ΓΘN ΕΥΑκκΉΑ±Υα≤–Μυ”κ C ΕΥΝρθΞΫχ––ΜΖΉ¥Ζ¥ ΫΝρθΞΜ·Θ§ΥφΚσΒΡ S ΒΫ N θΘΜυΉΣ“Τ…ζ≥…Υυ–ηΒΡΡΎθΘΑΖΜ·≤ζΈοΓΘfΘ§“Μ÷÷Ω…»Ξ≥ΐΒΡ N ΕΥ―θ““ΜυΝρ¥ΦΝ§Ϋ””Π”Ο”ΎκΡΒΡΆΖΈ≤ΜΖΜ·Θ§Ά®ΙΐάύΥΤ”Ύ‘≠ΈΜΜ·―ßΝ§Ϋ”ΒΡΖ¥”ΠΆΨΨΕΘ§Έό–η N ΕΥΑκκΉΑ±Υα≤–ΜυΓΘgΘ§Ά®ΙΐΈόΚέ Staudinger Ν§Ϋ”≤Ώ¬‘ Βœ÷κΡΒΡΆΖΈ≤ΜΖΜ·ΓΘ
ΩΥάοΤφΚΆΉτΉτΡΨΩΣΖΔΝΥ“Μ÷÷άϊ”ΟκΡΝρΥαθΞΚΆ…ΘΗώ ‘ΦΝ–Έ≥…θΘΑΖΦϋΒΡ–ρΝ–Θ§ΗΟ–ρΝ–Ω…”Ο”ΎΈεκΡΚΆΝυκΡΒΡΜΖΜ·Ζ¥”ΠΓΘ”ΟΏΏύΛ¥Πάμ N ΕΥ Fmoc ±ΘΜΛΚΆ C ΕΥ 9-ήΧΦΉΜυΝρθΞκΡΘ§Μα ΆΖ≈≥ω“Μ÷÷ΑκΝρΥαθΞκΡΘ§ΗΟκΡ‘Ύ 0.005 M …ΘΗώ ‘ΦΝΘ®FmocΘ§9-ήΧΦΉ―θτ ΜυΘ©¥φ‘Ύœ¬ΖΔ…ζΜΖΜ·Ζ¥”ΠΓΘΖ¥”ΠΆ®Ιΐ”κ…ΘΗώ ‘ΦΝΒΡ≥θ Φ SNAr Ζ¥”Π…ζ≥…‘≠ΈΜΜν–‘ΝρθΞΫχ––Θ§»ΜΚσ±Μ N ΕΥΑ±Μυ≤ΕΜώΓΘΗΟΖΫΖ®”κ”Έάκτ»ΥαΚΆτ«Μυœύ»ίΓΘάΐ»γΘ§Ής’ΏΡήΙΜ¥”œύ”ΠΒΡ±ΘΜΛœΏ–‘κΡ≥ωΖΔΘ§“‘ 44% ΒΡΉή ’¬ Ζ÷»ΐ≤ΫΚœ≥…ΜΖ-(d-Glu-Ala-d-Val-Leu-d-Trp)Θ®ΆΦ 4bΘ©ΓΘΥΡκΡ≤Μ ”Ο”Ύ’β÷÷ΜΖΜ·≤Ώ¬‘Θ§“ρΈΣ¥Πάμ Fmoc-Ala-Trp-Gly-Phe-SFm ÷ΜΒΟΒΫΜΖΕΰΨέΧε≤ζΈοΘ§≤ζ¬ ΈΣ 40%Θ®SFmΘ§9-ήΧΦΉΝρθΞΘ©ΓΘ
Ά®Ιΐ≤ύΝ¥”κ≤ύΝ¥ΒΡΝ§Ϋ” Βœ÷κΡΒΡΖ÷Ή”ΡΎΈ»Ε®Μ·’β“ΜΖΫΖ®”…Ζ―άϊΩΥΥΙΦΑΤδΆ§ ¬¬ œ»Χα≥ωΓΘΫΪκΡœό÷Τ≥…¥σΜΖΒΡΉν±ψΫίΓΔΉν÷±Ϋ”ΒΡΖΫΖ®÷°“Μ «Ά®ΙΐΝ§Ϋ”ΝΫΗωΡΎ≤ΩΑκκΉΑ±ΥαΒΡΝρ¥ΦΜυΆ≈ΓΘ”…¥Υ–Έ≥…ΒΡΖ÷Ή”ΡΎΕΰΝρΦϋΡήΙΜΈ»Ε®κΡ÷–ΒΡΕΰΦΕΫαΙΙΜυ–ρΓΘ“Σ Βœ÷’β÷÷ΜΖΜ·Θ§–η“Σ“Μ÷÷―θΜ·ΦΝΘ®»γ¥σΤχ÷–ΒΡ―θΤχΓΔΒβΓΔΕΰΦΉΜυ―«μΩΜρΕΰΘ®2-ΏΝύΛΜυΘ©ΕΰΝρΜ·ΈοΘ©ΓΘ‘Ύ»ή“Κ÷–ΜρΙΧœύ…œΙΙΫ®œΏ–‘κΡ–ρΝ– ±Θ§ΝΫΗωΝρ¥ΦΜυΆ≈ΕΦ”ΟœύΆ§ΒΡΜυΆ≈ΖβΕΥΘ§’β–©ΜυΆ≈‘ΎΜΖΜ·÷°«ΑΩ…“‘’ΐΫΜ«–ΗνΓΘΆ®≥ΘΘ§Νρ¥ΦΜυΆ≈”ΟΡήΙΜΆ§ ±Ά―±ΘΜΛ≤Δ–Έ≥…ΕΰΝρΦϋΒΡ±ΘΜΛΜυΆ≈±ΘΜΛΓΘάΐ»γΘ§S-““θΘΑ±ΜυΦΉΜυΘ®AcmΘ©ΚΆS-»ΐ±ΫΦΉΜυΘ®TrtΘ©±ΘΜΛΜυΆ≈ΕΦΩ…“‘”ΟΖ÷Ή”Ββ»Ξ≥ΐΘ§…ζ≥…―«Μ«θΘΒβ÷–ΦδΧεΘ§ΗΟ÷–ΦδΧε“ΣΟ¥‘ΎΤγΜ·Ζ¥”Π÷–”κΝμ“ΜΗω―«Μ«θΘΒβΖ¥”ΠΘ§“ΣΟ¥±Μ±ΘΜΛΒΡΝρ¥ΦΜυΆ≈ΙΞΜςΘ§¥”Εχ‘≠ΈΜ…ζ≥…ΕΰΝρΦϋΓΘΆ®Ιΐ≤ύΝ¥Ν§Ϋ” Βœ÷ΫαΙΙΈ»Ε®Μ· «“Μ÷÷Ζ«≥Θ”––ßΒΡΖΫΖ®ΓΘ’β «“ΜΗωΜν‘ΨΒΡ―–ΨΩΝλ”ρΘ§ΒΪ≥§≥ωΝΥ±ΨΉέ ωΒΡΖΕΈßΓΘ
ΑκκΉΑ±ΥαΒΡέœΜυΙΌΡήΆ≈“≤Ω…”Ο”ΎΖ÷Ή”ΡΎΒΡέœΜυ-œ©ΧΰΖ¥”Πά¥…ζ≥…κΡ¥σΜΖΓΘΑ≤»ϊΥΙΦΑΤδΆ§ ¬63‘χ≤…”Ο’β÷÷≤ύΝ¥ΒΫκΡΈ≤ΒΡΜΖΜ·≤Ώ¬‘ΓΘ’β÷÷¥σΜΖΜ·…φΦΑκΡΝ¥ N ΕΥΑκκΉΑ±Υα≤–ΜυΒΡέœΜυ”κκΡΝ¥ C ΕΥάΒΑ±Υα≤–ΜυΒΡ Π≈-NH2 …œΒΡœ©±ϊ―θτ Μυ±ΘΜΛΜυΆ≈ΒΡΥΪΦϋΫχ––Ή‘”…ΜυΦ”≥…Θ®ΆΦ 4cΘ©ΓΘΗΟΜΖΜ·Ζ¥”Π‘ΎΙΧœύ÷ß≥÷Έο…œ–ßΙϊΉνΦ―Θ§ΒΪ“≤Ω…‘Ύ»ή“ΚœύΜ·―ß÷–Ϋχ––Θ§Υυ–η≈®Ε»ΫœΒΆΘ®0.002 MΘ©ΓΘ
N ΕΥΑκκΉΑ±Υα≤–Μυ…œ¥φ‘ΎΒΡ 1,2-Α±ΜυΝρ¥ΦΙΌΡήΆ≈“―±Μ”Π”Ο”Ύ“ΜœΒΝ–¥σΜΖΜ·≤Ώ¬‘ΓΘΤδ÷–“Μ÷÷ΖΫΖ®…φΦΑ”κ»©Ϋχ––Ζ÷Ή”ΡΎΥθΚœΘ§–Έ≥…Έ»Ε®ΒΡύγΏρΏχ‘”ΜΖΓΘΧΖΦΑΤδΆ§ ¬÷ΛΟςΘ§Κ§”– N ΕΥΑκκΉΑ±Υα≤–ΜυΚΆΝ§Ϋ”άΒΑ±Υα≤–Μυ≤ύΝ¥ΒΡ»©ΙΌΡήΆ≈ΒΡΈ¥±ΘΜΛœΏ–‘κΡ»ί“ΉΥθΚœΘ§¥”ΕχΒΟΒΫ«Ε»κύγΏρΏχΜΖΒΡ≤ύΝ¥ΒΫΈ≤≤ΩΒΡΜΖκΡΓΘ¥ΥΆβΘ§ΥϊΟ«ΜΙΡήΙΜ÷ΛΟςΘ§»γΙϊΫΪ»©ΉςΈΣ―θΜ·ΒΡ C ΕΥΗ ”ΆθΞ≤Δ»κœΏ–‘«ΑΧε÷–Θ§Ρ«Ο¥Ά®Ιΐ“Μ÷÷–¬”±ΒΡΜΖ ’ΥθΜζ÷ΤΘ§Φ¥Ω…ΜώΒΟΨΏ”–»ΪθΘΑΖΦϋΙ«ΦήΒΡΆΖΒΫΈ≤ΜΖκΡΓΘΗΟΜζ÷ΤΆ®Ιΐ»ΐΜΖΖ÷Ή”ΡΎ÷Ί≈≈Ϋχ––Θ®ΆΦ 4dΘ©ΓΘ
1,2-Α±ΜυΝρ¥Φ‘ΎΒ±¥ζ“άάΒ≤ΕΜώ/÷Ί≈≈Μζ÷ΤΫΪΝΫΗωκΡΤ§ΕΈΝ§Ϋ”‘Ύ“ΜΤπΒΡΝ§Ϋ”≤Ώ¬‘÷–“≤»ΓΒΟΝΥΨό¥σ≥…ΙΠΓΘΧΊ±π «‘≠ΈΜΜ·―ßΝ§Ϋ”…ζΕ·ΒΊ’Ι ΨΝΥ’β÷÷Μζ÷Τ‘ΎΈ¬ΚΆΧθΦΰœ¬Ν§Ϋ”κΡΤ§ΕΈΒΡ«Ω¥σΡήΝΠΓΘΗΟΙΐ≥Χ…φΦΑΝΫΗωΤ§ΕΈ÷°ΦδΒΡΖ¥”ΠΘ§Τδ÷–“ΜΗωΤ§ΕΈ «»θΜνΜ·ΒΡ C ΕΥΝρθΞΘ§Νμ“ΜΗω «Έ¥±ΘΜΛΒΡ N ΕΥΑκκΉΑ±Υα≤–ΜυΓΘθΘΑΖΦϋœύΕ‘”ΎΝρθΞΒΡ»»ΝΠ―ß«ΩΕ» «ΗΟΖ¥”ΠΒΡ«ΐΕ·ΝΠΘ§Ά®ΙΐΝΎΫϋ«ΐΕ·ΒΡ S ΒΫ N θΘΜυ«®“ΤΒΟ“‘ Βœ÷ΓΘTam ΚΆ Pallin ΡήΙΜ÷ΛΟςΘ§Κ§”– N ΕΥΑκκΉΑ±Υα≤–ΜυΒΡ C ΕΥκΡΝρθΞ»ί“Ή“‘ΆΖΈ≤ΖΫ ΫΜΖΜ·Θ®ΆΦ 4eΘ©ΓΘ’β÷÷¥σΜΖΜ·Ω…“‘‘Ύ¥φ‘ΎΨΚ’υ–‘≤ύΝ¥ΙΌΡήΆ≈Θ®»γάΒΑ±ΥαΒΡ Π≈-NH2 ΜυΆ≈ΓΔΡΎ≤ΩΑκκΉΑ±ΥαΒΡΝρ¥ΦΜρΉιΑ±ΥαΒΡΏδΏρΘ©ΒΡ«ιΩωœ¬Ϋχ––ΓΘ‘ΎΜΖΝ¥ΜΞ±δ“λΙΙΤΫΚβΒΡΩΊ÷Τœ¬Θ§≥ΛΕ»¥” 5 ΒΫ 26 ΗωΑ±ΜυΥα≤–ΜυΒΡκΡΡήΙΜΜΖΜ·Θ§Ϋω≤ζ…ζΈΔΝΩΒΡΒΆΨέΈο≤ζΈοΓΘ
γ―ΕϊΚΆΩ®¬μάΉ¬ό±®ΒάΝΥ“Μ÷÷Χλ»ΜΜ·―ßΙ«ΦήΜΖΜ·Ζ¥”ΠΘ§ΗΟΖ¥”Π…φΦΑ“ΜΗωΆξ»ΪΆ―±ΘΜΛΒΡ 15 Ηω≤–ΜυΒΡκΡΘ§Τδ N ΕΥΈΣΑκκΉΑ±ΥαΘ§C ΕΥΈΣΝρθΞΓΘ‘ΎΗΏœΓ ΆΕ»Θ®0.0005 MΘ©ΚΆΫ”Ϋϋ÷––‘ pH ÷ΒΒΡΧθΦΰœ¬Θ§ΆΖΈ≤ΜΖΜ·Ζ¥”ΠΥ≥άϊΫχ––ΓΘ‘ΎΜΖΜ·≤Ϋ÷η÷–Ά§ ±Φ”»κΝΥ PhSH ΚΆ BnSHΓΘ’β–© ‘ΦΝΉςΈΣ«ΉΚΥ¥ΏΜ·ΦΝΘ§ ΙΖ÷Ή”ΦδΉΣΝρθΞΜ·ΆΨΨΕΒΟ“‘ Βœ÷Θ§¥”Εχ…ζ≥…“ΜΗωΗϋΨΏ«ΉΒγ–‘ΒΡ C ΕΥΜν–‘ΝρθΞΚΆ“ΜΗω‘ΎΑκκΉΑ±ΥαΝρ¥ΦΙΞΜςτ ΜυΚσΗϋ“Ήάκ»ΞΒΡΜυΆ≈ΓΘΧλ»ΜΜ·―ßΝ§Ϋ”ΒΡ–≠“ι“≤“―ά©’ΙΒΫΙΧœύ¥σΜΖΜ·Μ·―ßΝλ”ρΓΘ
Βά…≠‘ΎΚœ≥…ΒΑΑΉ÷ ΜΖΝ¥ ±…θ÷Ν Ι”ΟΝΥΧλ»ΜΜ·―ßΝ§Ϋ”Ζ® Βœ÷¥σΜΖΜ·Θ§Τδ÷–ΝΫΗωΜΖκΡœύΜΞΫΜ÷·‘Ύ“ΜΤπΓΘΥΰΡΖΦΑΤδΆ§ ¬Χα≥ωΒΡΓΑΝρά≠Ν¥Ζ¥”ΠΓ±“≤“‘ S ΒΫ N ΒΡθΘΜυ«®“ΤΈΣΚΥ–ΡΘΜΗΟ≤Ώ¬‘“―”Π”Ο”ΎΗΜΚ§ΑκκΉΑ±Υα≤–ΜυΒΡ¥σκΡΒΡΕΥΒΫΕΥΜΖΜ·ΓΘ¥ΥΦΕΝΣΖ¥”ΠΥυ–ηΒΡΙΊΦϋΙΌΡήΆ≈Αϋά®κΡΝ¥ N ΕΥΒΡΑκκΉΑ±ΥαΓΔ“ΜΗωΝρθΞ“‘ΦΑ÷Ν…Ό“ΜΗω«Ε»κκΡΝ¥ΡΎ≤ΩΒΡ”ΈάκέœΜυΓΘ
ΨΓΙή…œ ωΧαΒΫΒΡ”… S ΒΫ N ΒΡΫϋΨύ«ΐΕ·θΘΜυ«®“Τ¥σΜΖΜ·≤Ώ¬‘ °Ζ÷”––ßΘ§ΒΪΤδ÷ς“Σ»±œί‘Ύ”Ύ N ΕΥ±Ί–κ”–ΑκκΉΑ±Υα≤–ΜυΘ§’βœό÷ΤΝΥΤδΚœ≥…”ΟΆΨΓΘΈΣΝΥΩΥΖΰ’β“ΜΫαΙΙ“Σ«σΘ§ΩœΧΊΦΑΤδΆ§ ¬ΡήΙΜΆ®Ιΐ‘Ύ N ΕΥΑ±ΜυΥαΒΡΠΝ-Α±Μυ…œΝ§Ϋ”“ΜΗω―θ““Νρ¥ΦΜυΆ≈ά¥ΡΘΡβ N ΕΥΑκκΉΑ±ΥαΒΡ≤ύΝ¥ ΓΘΥϊΟ«ΡήΙΜ”––ßΒΊΫχ––‘≠ΈΜΜ·―ßΜΖΜ·Ζ¥”ΠΘ§”κ C ΕΥΝρθΞΫαΚœΘ§Κœ≥…≤ΜΚ§ΑκκΉΑ±Υα≤–ΜυΒΡΆΖΈ≤ΜΖκΡΘ§«“Έ¥Φλ≤βΒΫΜΖΒΆΨέΈοΓΘ‘Ύ S ΒΫ N ΒΡθΘΜυ«®“ΤΚσΘ§N-ΠΝ-―θ““Νρ¥ΦΜυΆ≈Ω…“‘Ά®Ιΐ‘ΎœΓ¥ΉΥα÷–”Ο–ΩΖέΜΙ‘≠»Ξ≥ΐΘ§¥”ΕχΒΟΒΫΜΖκΡΒΡΧλ»ΜΙ«ΦήΫαΙΙΘ®ΆΦ 4fΘ©ΓΘΉνΫϋΘ§Βά…≠ΚΆ―œΩΣΖΔΝΥ“Μ÷÷Ηϋ Β”ΟΒΡΖΫΖ®Θ§Ά®Ιΐ‘≠ΈΜΜ·―ßΝ§Ϋ”Ζ®ά¥ΜΖΜ·≤ΜΚ§ΑκκΉΑ±Υα≤–ΜυΒΡκΡΓΘ‘Ύ≥θ ΦΒΡ‘≠ΈΜΜ·―ߥσΜΖΜ·Ζ¥”Π÷–Θ§N ΕΥΑκκΉΑ±Υα≤–Μυ”κ C ΕΥΝρθΞΫαΚœΘ§ΗΟΜΖκΡΩ…Ά®Ιΐ”κάΉΡαΡχΫχ––Ά―ΝρΖ¥”ΠΕχ ß»ΞΤδΑκκΉΑ±Υα÷–ΒΡΝρΓΘ
ΙΰΩœ≤°ΗώΚΆΩΥά≥“ρΈ§…αΒ¬ΩΣΖΔΝΥ“Μ÷÷Μυ”ΎΝρθΞΒΡΜΖΜ·≤Ώ¬‘Θ§ΤδΖΫΖ®Μυ”Ύ“Μ÷÷ΈόΚέΒΡ ©Χ’ΕΓΗώΝ§Ϋ”Θ®ΆΦ 4gΘ©ΓΘ‘ΎκΡΒΡ C Ρ©ΕΥ”κΝρθΞœύΝ§ΒΡλΔ”κΈΜ”Ύ N Ρ©ΕΥΒΡΒΰΒΣΜ·ΈοΖΔ…ζΖ÷Ή”ΡΎΖ¥”ΠΘ§–Έ≥…ΜΖΉ¥―«ΑΖΝΉΆιΓΘΥφΚσΘ§ΗΟΜΖ ’Υθ–Έ≥…θΘΑΖΦϋΘ§“ρΈΣΒΣ‘”“ΕΝΔΒ¬ΙΞΜςΝΥΝρθΞΒΡ«ΉΒγ÷––ΡΓΘ’β÷÷¥σΜΖΡΎθΘΑΖΜ·Ιΐ≥Χ“―±Μ÷ΛΟς ”Ο”ΎΦΗ÷÷Άξ»ΪΆ―±ΘΜΛΒΡκΡΒΡΜΖΜ·Θ§’β–©κΡ”… 11 ΗωΑ±ΜυΥα≤–ΜυΉι≥…ΓΘ’βΈΣ÷±Ϋ”ΜώΒΟΜΖκΡΧαΙ©ΝΥΆΨΨΕΘ§Έό–ηΫχ“Μ≤ΫΒΡΆ―±ΘΜΛ≤Ϋ÷ηΓΘ
…φΦΑΡΎθΞΒΡΜΖ ’Υθ≤Ώ¬‘
Ά®Ιΐ–Έ≥…“ΜΗωΗϋ¥σΓΔΗϋΝιΜνΒΡ¥σΜΖΘ§»ΜΚσΆ®ΙΐΖ÷Ή”ΡΎΜΖ ’Υθά¥ΜώΒΟΥυ–ηΒΡΡΩ±ξΜ·ΚœΈοΘ§’β «Φθ«α¥σΜΖΜ·λΊΥπ ßΒΡ”––ßΖΫΖ®ΓΘΑ≤≤Φά≠Β¬ΦΑΤδΆ§ ¬άϊ”Ο O ΒΫ N «®“Τ≤Ώ¬‘Κœ≥…ΝΥΜΖκΡΓΘΑ≤≤Φά≠Β¬ΒΡ≤Ώ¬‘Μυ”Ύ Ήœ»Ε‘ΙΙœσΝιΜνΒΡκΡθΞΫχ––¥σΜΖΜ·Θ§»ΜΚσΫχ–– O ΒΫ N θΘΜυ«®“Τ“‘…ζ≥…Ά§‘¥ΜΖκΡΘ®ΆΦ 5aΘ©ΓΘκΡ–ρΝ– Ήœ»¥”ΙΧΕ®‘ΎΙΧœύ‘ΊΧε…œΒΡ N-Boc ±ΘΜΛΒΡΥΩΑ±Υα≤ύΝ¥ΙΙΫ®Θ§–Έ≥… O-θΘΜυ“λκΡΦϋΘ®BocΘ§ εΕΓ―θτ ΜυΘ©ΓΘ¥”ΙΧœύ‘ΊΧε…œ«–Ηνœ¬ά¥ΚσΘ§‘ΎΗΏœΓ ΆΧθΦΰœ¬ΫΪκΡθΞΜΖΜ·Θ§…ζ≥…œύ”ΠΒΡΜΖΉ¥άύΥΤΈοΓΘ»Ξ≥ΐΥΩΑ±Υα≤–ΜυΒΡ N-Boc ±ΘΜΛΜυΚσΘ§‘ΎΦν–‘ΧθΦΰœ¬Ϋχ––Ήν÷’ΒΡ O ΒΫ N θΘΜυ«®“ΤΘ§“‘÷–Β»≤ζ¬ …ζ≥…Χλ»ΜΜΖκΡΓΘΗΟ≤Ώ¬‘“―÷ΛΟς ”Ο”ΎΜΖΈεκΡΚΆΜΖΝυκΡΒΡΚœ≥…Θ§»ΜΕχΑ≤≤Φά≠Β¬ΦΑΤδΆ§ ¬Ιέ≤λΒΫΕ‘”ΎΙΙœσΗϋ ήœό÷ΤΒΡΥΡκΡ―ή…ζΈοΘ§θΘΜυ«®“Τ≤Ϋ÷η≤ΔΈ¥Ϋχ––ΓΘ
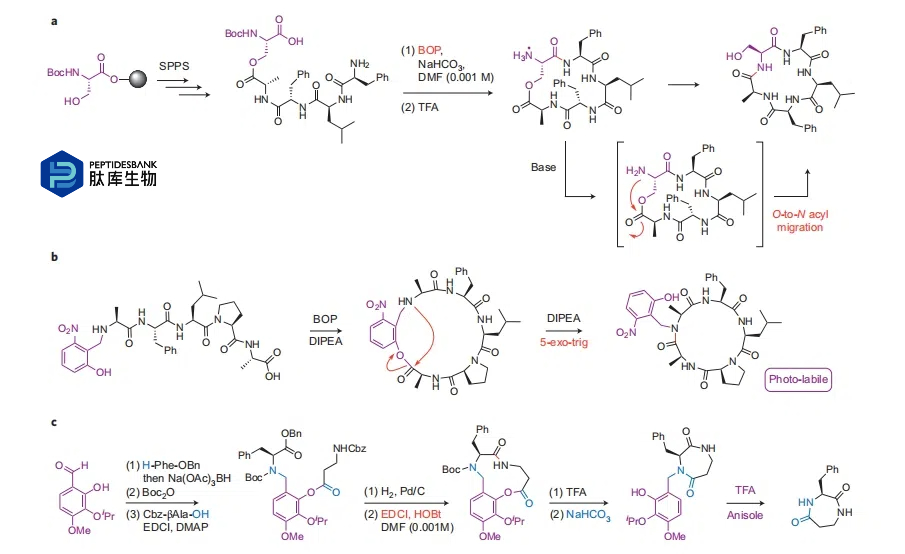 ΆΦ 5 | Ά®Ιΐ…φΦΑΫœ¥σΡΎθΞΜΖΒΡΜΖ ’Υθ Βœ÷κΡΒΡΡΎθΘΑΖΜ·ΓΘaΘ§“Μ÷÷ΜΖ ’Υθ≤Ώ¬‘Θ§Ά®ΙΐΫΪ“ΜΗωκΡθΞΒΡθΘΜυ¥” O “Τ÷Ν N “‘…ζ≥…Ά§‘¥ΈεκΡΜΖκΡΓΘ Ήœ»¥”ΥΩΑ±Υα≤–ΜυΒΡ≤ύΝ¥ΙΙΫ®œΏ–‘«ΑΧεΓΘ¥”ΙΧœύ‘ΊΧε…œ«–Ηνœ¬ά¥ΚσΘ§Ϋχ––¥σΜΖΡΎθΘΑΖΜ·“‘…ζ≥…κΡθΞΓΘΥφΚσΫχ–– Boc ±ΘΜΛΜυΆ―≥ΐΘ§ ΆΖ≈≥ωΥΩΑ±Υα≤–ΜυΒΡΠΝ-Α±ΜυΘ§¥”ΕχΖΔ…ζθΘΜυΉΣ“ΤΖ¥”ΠΓΘbΘ§“Μ÷÷ΙβΩ…Ν―ΫβΗ®÷ζΜυΆ≈ HnB ”Ο”ΎΜΖΜ·ΨΏ”–Χτ’Ϋ–‘ΒΡΈεκΡ H-Ala-Phe-Leu-Pro-Ala-OH ΒΡΜΖ ’Υθ≤Ώ¬‘ΓΘΫΪΗΟΗ®÷ζΜυΆ≈“ΐ»κκΡΒΡ N ΕΥΚσΘ§ΜΖΜ·Ά®Ιΐ≥θ ΦΒΡΜΖΉ¥œθΜυ±ΫθΞΫχ––Θ§ΗΟθΞ‘Λœ»Ήι÷·κΡΫχ––ΡΎθΘΑΖΜ·Θ§Ά®ΙΐΖ÷Ή”ΡΎ O ΒΫ N ΒΡθΘΜυΉΣ“Τ…ζ≥…Ά§‘¥ΈεκΡΜΖκΡΓΘcΘ§“Μ÷÷”…Υ°―ν»©―ή…ζΒΡΗ®÷ζΜυΆ≈Θ§ Ήœ»ΉςΈΣκΡ¥σΜΖΡΎθΘΑΖΜ·ΒΡΝιΜνΫ¬Ν¥ΓΘΥφΚσΫχ–– Boc ±ΘΜΛΜυΆ―≥ΐΘ§¥σΜΖ‘ΎΦν–‘ΧθΦΰœ¬Ά®ΙΐΜΖ ’ΥθΫχ–– O ΒΫ N ΒΡθΘΜυΉΣ“ΤΓΘ»ΜΚσ‘ΎΥα–‘ΧθΦΰœ¬ΫΪΈΜ“ΤΒΡΗ®÷ζΜυΆ≈Ν―ΫβΘ§¥”ΕχΒΟΒΫΥυ–ηΒΡΆ§ΕΰΆΣΏΏύΚΓΘEDCIΘ§1-““Μυ-3-(3-ΕΰΦΉΑ±Μυ±ϊΜυ)ΧΦΕΰ―«ΑΖ―ΈΥα―Έ
ΆΦ 5 | Ά®Ιΐ…φΦΑΫœ¥σΡΎθΞΜΖΒΡΜΖ ’Υθ Βœ÷κΡΒΡΡΎθΘΑΖΜ·ΓΘaΘ§“Μ÷÷ΜΖ ’Υθ≤Ώ¬‘Θ§Ά®ΙΐΫΪ“ΜΗωκΡθΞΒΡθΘΜυ¥” O “Τ÷Ν N “‘…ζ≥…Ά§‘¥ΈεκΡΜΖκΡΓΘ Ήœ»¥”ΥΩΑ±Υα≤–ΜυΒΡ≤ύΝ¥ΙΙΫ®œΏ–‘«ΑΧεΓΘ¥”ΙΧœύ‘ΊΧε…œ«–Ηνœ¬ά¥ΚσΘ§Ϋχ––¥σΜΖΡΎθΘΑΖΜ·“‘…ζ≥…κΡθΞΓΘΥφΚσΫχ–– Boc ±ΘΜΛΜυΆ―≥ΐΘ§ ΆΖ≈≥ωΥΩΑ±Υα≤–ΜυΒΡΠΝ-Α±ΜυΘ§¥”ΕχΖΔ…ζθΘΜυΉΣ“ΤΖ¥”ΠΓΘbΘ§“Μ÷÷ΙβΩ…Ν―ΫβΗ®÷ζΜυΆ≈ HnB ”Ο”ΎΜΖΜ·ΨΏ”–Χτ’Ϋ–‘ΒΡΈεκΡ H-Ala-Phe-Leu-Pro-Ala-OH ΒΡΜΖ ’Υθ≤Ώ¬‘ΓΘΫΪΗΟΗ®÷ζΜυΆ≈“ΐ»κκΡΒΡ N ΕΥΚσΘ§ΜΖΜ·Ά®Ιΐ≥θ ΦΒΡΜΖΉ¥œθΜυ±ΫθΞΫχ––Θ§ΗΟθΞ‘Λœ»Ήι÷·κΡΫχ––ΡΎθΘΑΖΜ·Θ§Ά®ΙΐΖ÷Ή”ΡΎ O ΒΫ N ΒΡθΘΜυΉΣ“Τ…ζ≥…Ά§‘¥ΈεκΡΜΖκΡΓΘcΘ§“Μ÷÷”…Υ°―ν»©―ή…ζΒΡΗ®÷ζΜυΆ≈Θ§ Ήœ»ΉςΈΣκΡ¥σΜΖΡΎθΘΑΖΜ·ΒΡΝιΜνΫ¬Ν¥ΓΘΥφΚσΫχ–– Boc ±ΘΜΛΜυΆ―≥ΐΘ§¥σΜΖ‘ΎΦν–‘ΧθΦΰœ¬Ά®ΙΐΜΖ ’ΥθΫχ–– O ΒΫ N ΒΡθΘΜυΉΣ“ΤΓΘ»ΜΚσ‘ΎΥα–‘ΧθΦΰœ¬ΫΪΈΜ“ΤΒΡΗ®÷ζΜυΆ≈Ν―ΫβΘ§¥”ΕχΒΟΒΫΥυ–ηΒΡΆ§ΕΰΆΣΏΏύΚΓΘEDCIΘ§1-““Μυ-3-(3-ΕΰΦΉΑ±Μυ±ϊΜυ)ΧΦΕΰ―«ΑΖ―ΈΥα―Έ
MeutermansΓΔSmythe ΦΑΤδΆ§ ¬ΩΣΖΔΝΥ“Μ÷÷…φΦΑΡΎθΞΒΡΝμ“Μ÷÷ΜΖ ’Υθ≤Ώ¬‘Θ§≤ΔΫΪΤδ”Π”Ο”Ύ–ΓκΡΒΡΆΖΈ≤ΜΖΜ·Ζ¥”ΠΓΘΆ®ΙΐΜΙ‘≠ΑΖΜ·Θ§ΫΪ“Μ÷÷Υ°―ν»©―ή…ζΒΡΗ®÷ζΜυΆ≈Ν§Ϋ”ΒΫκΡΝ¥ΒΡ N ΕΥΓΘC ΕΥΒΡΜνΜ·Ήν≥θΒΦ÷¬–Έ≥…Ηϋ»ί“ΉΫ”ΫϋΒΡΡΎθΞΓΘ’β Ι N ΕΥΩΩΫϋ C ΕΥΘ§≤Δ¥ΌΫχΝΥ O ΒΫ N ΒΡθΘΜυΉΣ“ΤΘ®ΆΦ 5bΘ©ΓΘ’β÷÷ΜνΜ·Α±ΜυΥαΒΡθΘΜυ≤ΕΜώΖΫΖ®ΥφΚσ”κΜυ”Ύ 2-τ«Μυ±ΫΦΉΜυΒΡΗ®÷ζΜυΆ≈Ϋχ––θΘΜυΉΣ“ΤΘ§Ήν≥θ”… Sheppard ΦΑΤδΆ§ ¬ΩΣΖΔΘ§ΉςΈΣ Βœ÷ ήΉηκΡθΘΑΖ÷ςΝ¥»Γ¥ζΒΡ”––ßΖΫΖ®76ΓΘSmythe ΦΑΤδΆ§ ¬―Γ‘ώ 2-τ«Μυ-6-œθΜυ±ΫΦΉΜυΘ®HnBΘ©ΉςΈΣΗ®÷ζΜυΆ≈Θ§“ρΈΣΥϋΨΏ”–Ιβ≤ΜΈ»Ε®–‘Θ§‘Ύ¥σΜΖΜ·ΚσΩ…“‘»Ξ≥ΐΓΘΉςΈΣΗ≈Ρν―ι÷ΛΘ§Ρ―“‘ΜΖΜ·ΒΡΈεκΡ H-Ala-Phe-Leu-Pro-Ala-OHΘ®ΆΦ 1dΘ©”κ HnB Ν§Ϋ”Θ§‘Ύ 0.001 M ≈®Ε»œ¬“‘ 45% ΒΡ≤ζ¬ ΜΖΜ·ΓΘΉς’ΏΜΙ”Π”Ο’β÷÷ΜΖ ’Υθ≤Ώ¬‘Κœ≥…ΝΥΜΖΥΡκΡΩβΓΘΥϊΟ«…θ÷ΝΡήΙΜ÷ΛΟςΘ§’β÷÷≤Ώ¬‘ ”Ο”ΎΚœ≥…ΗΏΕ» ήœόΒΡ»Ϊ L –ΆΜΖΉ¥ΥΡκΡΓΘ»γΙϊ‘ΎΤδ÷–“ΜΗω÷ςΝ¥θΘΑΖΒΣ‘≠Ή”…œΑ≤ΉΑ“ΜΗωΕνΆβΒΡ HnB ΒΞ‘ΣΘ§“‘”ΑœλΥ≥ ΫθΘΑΖΉΣΫ«ΫαΙΙΘ§¥”Εχ”–÷ζ”ΎΫΪΡ©ΕΥά≠ΒΟΗϋΫϋΘ§Ρ«Ο¥’β÷÷άßΡ―ΒΡΜΖΜ·Ζ¥”Π «Ω…––ΒΡΓΘ
ΖΕΓΛ¬μΕϊ»ϊΈΡΦΑΤδΆ§ ¬ΩΣΖΔΝΥ“Μ÷÷”Ο”ΎάßΡ―¥σΜΖΡΎθΘΑΖΜ·ΒΡ«·–ΈΗ®÷ζ≤Ώ¬‘ΓΘ‘ΎΥϊΟ«ΒΡΖΫΖ®÷–Θ§ΫΪΥ°―ν»©―ή…ζΒΡΗ®÷ζΜυΆ≈“ΐ»κœΏ–‘κΡΒΡ÷ςΝ¥÷–Θ§–Έ≥…“ΜΗωΝιΜνΒΡΝ§Ϋ”ΧεΜρΓΑΫ¬Ν¥Γ±Θ§¥”Εχ Βœ÷ΆΖΈ≤¥σΜΖΡΎθΘΑΖΜ·ΓΘΥφΚσΒΡΜΖ ’Υθ…φΦΑ“ΜΗω O ΒΫ N ΒΡθΘΜυΉΣ“ΤΖ¥”ΠΘ®ΆΦ 5cΘ©ΓΘ‘Ύ’βάοΘ§Υ°―ν»©―ή…ζΒΡΗ®÷ζΜυΆ≈ΉςΈΣ¥σΜΖΡΎθΘΑΖΜ·ΒΡΡΘΑεΘ§Τδ≥…ΙΠ÷°¥Π‘Ύ”ΎΝΫΖΫΟφΓΘ Ήœ»Θ§”…”ΎΖΦΜυθΞΕ‘Α±ΫβΒΡΗΏΖ¥”Π–‘Θ§¥χά¥ΝΥλ ΦΛΜνΓΘΤδ¥ΈΘ§Η®÷ζΜυΆ≈’ΐ»ΖΒΊΫΪ÷ΌΑΖΚΆτ ΜυΙΌΡήΆ≈Ε®ΈΜ‘ΎΜΖ ’Υθ ±ΒΡΫϋΝΎΈΜ÷ΟΘ§¥”Εχ¥χά¥ΝΥλΊΦΛΜνΓΘΗΟ≤Ώ¬‘“―≥…ΙΠ”Π”Ο”ΎΦΗ÷÷Ρ―“‘Κœ≥…ΒΡΆ§ΕΰΆΣΏΏύΚΒΡΚœ≥…ΓΘ
ΒΰΒΣΜ·Έο-»≤ΧΰΜΖΦ”≥…Ζ¥”Π
¥”ΚΘ―σ…ζΈο÷–Ζ÷άκ≥ωΒΡ¥σΝΩΨΏ”–…ζΈοΜν–‘ΒΡΫαΙΙΑϋΚ§”–«Ε»κΫœ–Γ‘”ΜΖΒΡΜΖκΡΓΘ‘Ύ“Μ–©άΐΉ”÷–Θ§ύγΏρΚΆΕώΏρΜΖΒΡ¥φ‘ΎΕ‘œύ”ΠΒΡ¥σΜΖ ©Φ”ΝΥΙΙœσœό÷ΤΓΘ“ρ¥ΥΘ§ΫΪ‘”ΜΖ“ΐ»κΜΖκΡΒΡ÷ςΝ¥‘Ϋά¥‘Ϋ ήΒΫΙΊΉΔΘ§”»Τδ «‘Ύ“©ΈοΜ·―ßΝλ”ρΓΘ’β
1,2,3-»ΐΏρ «“Μ÷÷ΧΊ±π“ΐ»ΥΙΊΉΔΒΡ‘”ΜΖΜ·ΚœΈοΘ§“ρΈΣΥϋ‘Ύ»»ΝΠ―ßΚΆ…ζάμ―ß…œΕΦΚήΈ»Ε®ΓΘΗυΨίΤδ»Γ¥ζΡΘ ΫΘ§ΥϋΩ…“‘ΉςΈΣΖ¥ ΫΜρΥ≥ ΫθΘΑΖΦϋΒΡ”––ßΒ»ΒγΉ”ΧεΓΘ’ΐ»γ Huisgen Ήν≥θΥυ’Ι ΨΒΡΡ«―υΘ§’β÷÷‘”ΜΖΜ·ΚœΈοΩ…“‘Ά®ΙΐΒΰΒΣΜ·ΈοΚΆ»≤Χΰ÷°ΦδΒΡ 1,3-≈ΦΦΪΜΖΦ”≥…Ζ¥”Π«αΥ…ΜώΒΟΓΘΉνΫϋΘ§Meldal ΚΆ Sharpless ΒΡ―–ΨΩ–ΓΉιΩΣΖΔΝΥ“Μ÷÷‘ΎΈ¬ΚΆΧθΦΰœ¬”…Ά≠Θ®IΘ©¥ΏΜ·ΒΡΗΏΕ»«χ”ρ―Γ‘ώ–‘±δΧεΘ§“‘…ζ≥… 1Θ§4-Εΰ»Γ¥ζΡΘ ΫΓΘ’β÷÷ΓΑΒψΜςΓ±Μ·―ߥΥΚσ“―≥…ΙΠ”Π”Ο”ΎκΡΒΡΜΖΜ·ΓΘ
÷°«ΑΈόΖ®ΜώΒΟΒΡΜΖΉ¥[Pro-Val-Pro-Tyr]10Θ®ΆΦ1cΘ©ΒΡ»ΐΏράύΥΤΈοΩ…“‘Ά®ΙΐNΕΥΒΰΒΣΜ·ΈοΚΆCΕΥ»≤ΧΰΒΡΜΖΦ”≥…Ζ¥”Π«αΥ…Κœ≥…Θ®ΆΦ6aΘ©ΓΘ÷Ί“ΣΒΡ «Θ§’β–©Β»≈≈Χε±Μ÷ΛΟς±ΘΝτΝΥΤδά“Α±ΥαΟΗ“÷÷ΤΜν–‘ΓΘ
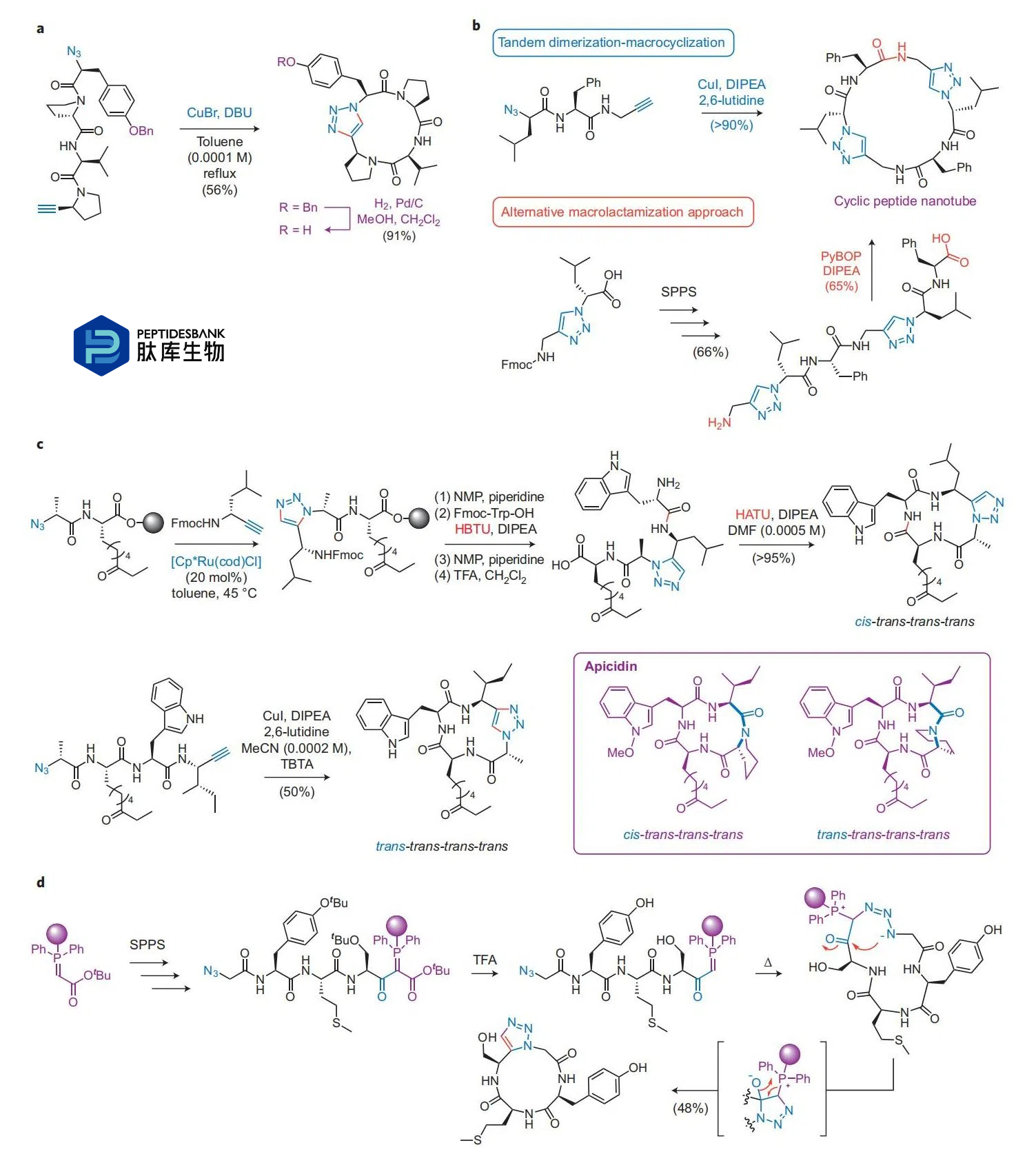
ΆΦ 6 | κΡ¥σΜΖΚœ≥…÷–ΒΡΒΰΒΣ - »≤ΧΰΜΖΦ”≥…Ζ¥”ΠΓΘaΘ§ά“Α±Υα - ±ϊΑ±Υα - γ”Α±Υα - ±ϊΑ±ΥαΒΡΒψΜςΫιΒΦ¥σΜΖΜ·ΓΘbΘ§Ά®ΙΐΝΫ÷÷ΒΰΒΣ - »≤ΧΰΒψΜςΖ¥”ΠΒΡ¥°ΝΣΕΰΨέ - ¥σΜΖΜ·ΖΫΖ®Θ®…œΘ©ΜρΆ®Ιΐ–߬ ΫœΒΆΒΡ¥ΪΆ≥¥σΜΖΡΎθΘΑΖΜ·ΖΫΖ®Θ®œ¬Θ©Κœ≥…ΜΖκΡΡ…ΟΉΙήΓΘcΘ§ΜΖΥΡκΡ apicidin ΒΡ»ΐΏρ–ό ΈάύΥΤΈοΒΡΚœ≥…ΓΘ…œΘΚ‘ΎΙΧœύ…œΆ®Ιΐν…¥ΏΜ·–Έ≥… 1Θ§5- Εΰ»Γ¥ζ 1Θ§2Θ§3- »ΐΏρΘ§ΥφΚσΫχ––¥σΜΖΡΎθΘΑΖΜ·Θ§ΒΟΒΫάύΥΤ”Ύ apicidin …ζΈοΜν–‘ΙΙœσΒΡάύΥΤΈοΓΘœ¬ΘΚΆ®ΙΐΆ≠Θ®ΔώΘ©¥ΏΜ·ΒΡΖ÷Ή”ΡΎΒΰΒΣ - »≤ΧΰΜΖΦ”≥…Ζ¥”ΠΘ§ΒΟΒΫάύΥΤ”Ύ apicidin ‘Ύ»ή“Κ÷–ΒΡ÷ς“ΣΙΙœσΒΡάύΥΤΈοΓΘdΘ§Ά®ΙΐΙΧœύΝ§Ϋ”ΒΡΒΰΒΣκΡΜυΝΉ“ΕΝΔΒ¬ΒΡΖ÷Ή”ΡΎΜΖΜ·Ν―ΫβΚœ≥…Κ§ 1Θ§5- Εΰ»Γ¥ζ 1Θ§2Θ§3- »ΐΏρΒΡΜΖΥΡκΡάύΥΤΈοΓΘHATUΘ§2-(7-ΒΣ‘” -1H- ±Ϋ≤Δ»ΐΏρ -1- Μυ) -1Θ§1Θ§3Θ§3- ΥΡΦΉΜυκεΝυΖζΝΉΥαθΞΘΜNMPΘ§N- ΦΉΜυ -2- ΏΝΩ©ΆιΆΣΘΜTBTAΘ§»ΐ[Θ®1-ή–Μυ-1H-1,2,3-»ΐΏρ-4-ΜυΘ©ΦΉΜυ]ΑΖΓΘ
Lokey ΦΑΤδΆ§ ¬Ϋχ“Μ≤Ϋ÷ΛΟςΝΥΗΟΖ¥”ΠΉςΈΣ¥σΜΖΜ·ΙΛΨΏΒΡ Β”Ο–‘ΓΘΥϊΟ«ΡήΙΜΕ‘ΗΜΚ§ΝΝΑ±ΥαΒΡΥΡκΡΓΔΈεκΡΓΔΝυκΡΚΆΤΏκΡΫχ––ΙΧœύ÷ß≥÷ΜΖΜ·ΓΘ‘Ύ–μΕύΆ≠¥ΏΜ·ΒΡΒΰΒΣΜ·Έο-»≤ΧΰΜΖΦ”≥…Ζ¥”Π÷–ΕΦΙέ≤λΒΫΝΥΕΰΨέΧεΚΆ»ΐΨέΧεΗ±≤ζΈοΒΡ–Έ≥…ΓΘGhadiri ΦΑΤδΆ§ ¬άϊ”ΟΝΥ’β“Μœ÷œσΘΜΆ®Ιΐ”…ΦΗΗω¥°ΝΣΒψΜςΖ¥”Π Βœ÷ΒΡ¥°ΝΣΕΰΨέΜ·-¥σΜΖΜ·ΖΫΖ®Θ§ΥϊΟ«ΡήΙΜΚœ≥… C2 Ε‘≥ΤΒΡΜΖκΡ÷ßΦήΘ§’β–©÷ßΦή±Μ÷ΛΟςΡήΙΜΉ‘ΉιΉΑ≥…κΡΡ…ΟΉΙήΓΘ’β÷÷ΖΫΖ®±»Ε‘“―Ψ≠ΑϋΚ§ΝΫΗω»ΐΏρΒΞ‘ΣΒΡœΏ–‘«ΑΧεΫχ––≥ΘΙφ¥σΜΖΡΎθΘΑΖΜ·»ΓΒΟΝΥΗϋ¥σΒΡ≥…ΙΠΘ®ΆΦ6bΘ©ΓΘ
1,4-Εΰ»Γ¥ζΒΡ 1,2,3-»ΐΏρ «“―÷ΣΒΡΖ¥ ΫθΘΑΖΦϋΒΡΧφ¥ζΈοΓΘΦ”ΒœάοΦΑΤδΆ§ ¬“―÷ΛΟςΘ§ΫΪ“ΜΗωΜρΝΫΗω¥ΥάύΜυΆ≈“ΐ»κ“―÷ΣΕ‘…ζ≥Λ“÷ΥΊΘ®SSTΘ© ήΧεΨΏ”–«ΩΫαΚœ«ΉΚΆΝΠΒΡΜΖΉ¥ΥΡκΡ÷–Θ§Ω……ζ≥…ΙΙœσΨυ“ΜΒΡάύΥΤΈοΓΘ ΒΦ …œΘ§’β–©Έ±ΥΡκΡΦ¥ Ι‘Ύ 70ΓψC ±“≤Ϋω≥ œ÷“Μ÷÷ΙΙœσΓΘ
ΝμΆβΘ§1,5-Εΰ»Γ¥ζΒΡ 1,2,3-»ΐΏρ“―±Μ÷ΛΟς «”––ßΒΡΥ≥ ΫθΘΑΖΦϋΧφ¥ζΈοΘ§Ρή”’ΒΦΉΣΫ«ΫαΙΙΜυ–ρΓΘ’β–©Μ·ΚœΈοΩ…“‘Ά®Ιΐν…Θ®IIΘ©¥ΏΜ·ΒΡΒΰΒΣΜ·ΈοΚΆ»≤ΧΰΒΡ 1,3-≈ΦΦΪΜΖΦ”≥…Ζ¥”Π“‘«χ”ρ―Γ‘ώ–‘ΒΡΖΫ Ϋ÷Τ±ΗΓΘΦ”ΒœάοΦΑΤδΆ§ ¬ΡήΙΜΫΪ’β–©‘”ΜΖΉςΈΣΥ≥ ΫθΘΑΖΒ»≈≈Χε“ΐ»κ“Μ÷÷Χλ»Μ¥φ‘ΎΒΡΜΖΥΡκΡ÷–ΓΘ’β «Ά®Ιΐ‘ΎΙΧœύ…œ Ήœ»Ϋχ––ν…Θ®IIΘ©¥ΏΜ·ΒΡ–Έ≥…Κ§œΏ–‘κΡΒΡ 1,5-Εΰ»Γ¥ζ 1,2,3-»ΐΏρΘ§»ΜΚσΕ‘ΦΌΥΡκΡΫχ––≥ΘΙφΒΡΜΖΚœΖ¥”Πά¥ Βœ÷ΒΡΘ®ΆΦ 6cΘ©ΓΘΆ®Ιΐ’β÷÷ΖΫ ΫΘ§ΥϊΟ«ΡήΙΜ÷Τ±Η≥ωΙΙœσΨυ“ΜΒΡΑΔΤΛΈςΕΓάύΥΤΈοΓΘΒδ–ΆΒΡΜΖΥΡκΡ‘Ύ»ή“Κ÷–≥ œ÷»ΪΖ¥ ΫθΘΑΖΫαΙΙΘ§»ΜΕχΤδ…ζΈοΜν–‘ΙΙœσΑϋΚ§“ΜΗωΥ≥ ΫθΘΑΖ–ΐΉΣ“λΙΙΧεΓΘ ΒΦ …œΘ§Ής’ΏΡήΙΜ÷ΛΟςΤδ“ΐ»κ 1,5-Εΰ»Γ¥ζ 1,2,3-»ΐΏρΒΡάύΥΤΈοœ‘ Ψ≥ω”κΧλ»Μ¥φ‘ΎΒΡΑΔΤΛΈςΕΓœύΥΤΒΡ…ζΈοΜν–‘ΓΘ
ΉνΫϋΘ§ά≠Β¬¬ϋΚΆΑΔ…Θ≈§ά≠≥…ΙΠ±®ΒάΝΥΒΰΒΣΜυ»≤ΜυκΡΒΡ Ή¥ΈΜΖΜ·Ζ¥”ΠΘ§¥”ΕχΚœ≥…ΝΥΚ§ 1,5-Εΰ»Γ¥ζ»ΐΏρΒΡΜΖκΡΓΘΥϊΟ«ΩΣΖΔΝΥ“Μ÷÷Μυ”ΎΙΧœύ÷ß≥÷≤Ώ¬‘ΒΡΜΖΜ·Ν―ΫβΖΫΖ®Θ§Τδ÷–ΜΖΜ·ΚΆ¥”ΙΧœύ÷ß≥÷…œΝ―Ϋβ‘ΎΆ§“ΜΗωΜ·―ßΖ¥”Π÷–Ϋχ––ΓΘ’β÷÷ΖΫΖ®±ψ”Ύ¥ΩΜ·Θ§“ρΈΣΩΣΝ¥Ι―ΨέΗ±≤ζΈο»‘ΗΫΉ≈‘ΎΙΧœύ÷ß≥÷…œΓΘΗΟΜ·―ßΖ¥”Π…φΦΑΨέΚœΈοΫαΚœΒΰΒΣΜυκΡΜυΝΉ“ΕΝΔΒ¬ΒΡΕΰΦΪΜΖΦ”≥…Θ§±ήΟβΝΥ Ι”ΟΑ±ΜυΥα»≤ΧΰΘ§≤Δ«“Έό–ηΫπ τΘ®ΆΦ 6dΘ©ΓΘ
ΜΖΚœΗ¥Ζ÷ΫβΖ¥”Π
‘Ύ–Έ≥…ΧΦ-ΧΦΦϋΖΫΟφΘ§œ©ΧΰΗ¥Ζ÷ΫβΖ¥”ΠΒΡ≥ωœ÷ΈΣ¥σΜΖΜ·Νλ”ρ¥χά¥ΝΥΙψΖΚΒΡ”Π”ΟΓΘΗώ¬≥≤ΦΥΙΦΑΤδΆ§ ¬ΩΣΖΔΒΡΗΏΕ»ΡΆ ήΙΌΡήΆ≈ΒΡν…Μυ¥ΏΜ·ΦΝΦΪ¥σΒΊ¥ΌΫχΝΥ”–ΜζΜ·―ß’β“ΜΝλ”ρœρκΡάύΦΑœύΙΊ…ζΈοΧεœΒΒΡΆΊ’ΙΓΘΗώ¬≥≤ΦΥΙΦΑΤδΆ§ ¬¬ œ»ΫΪΜΖΚœΗ¥Ζ÷ΫβΘ®RCMΘ©≤Ώ¬‘”Π”Ο”ΎΙΙœσΗ’–‘Μ·Α±ΜυΥαΚΆκΡάύ95ΓΘ ή“ΜάύΧλ»Μ¥φ‘ΎΒΡΓΔΆ®ΙΐΕΰΝρΦϋΈ»Ε®Μ·ΒΡΠ¬-ΉΣΫ«ΫαΙΙΜυ–ρΒΡΤτΖΔΘ§ΥϊΟ«‘Ύ’β–©ΥΡκΡ–ρΝ–÷–”ΟΧΦ-ΧΦΦϋ»Γ¥ζΝΥΕΰΝρΦϋΓΘ’β «Ά®Ιΐœ©±ϊΜυΗ Α±Υα≤–Μυ÷°ΦδΒΡ RCM Ζ¥”Π Βœ÷ΒΡΘ§≤Δ«“ΖΔœ÷ΗΟΖ¥”ΠΡήΙΜΡΆ ήΥα–‘θΘΑΖ NH ÷ Ή”“‘ΦΑΈ¥±ΘΜΛΒΡά“Α±ΥαΖ”ΜυΘ®ΆΦ 7aΘ©ΓΘ
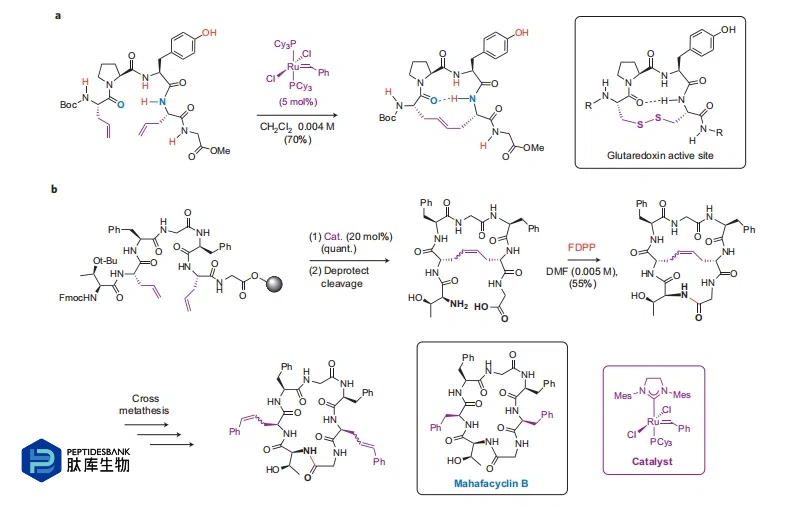
ΆΦ 7 | ΜΖΚœΗ¥Ζ÷ΫβΖ¥”Π‘ΎκΡ¥σΜΖΚœ≥…÷–ΒΡ”Π”ΟΓΘaΘ§Ά®ΙΐΜΖΚœΗ¥Ζ÷ΫβΖ¥”Π Βœ÷≤ύΝ¥ΒΫ≤ύΝ¥ΒΡΜΖΜ·≤ΔΈ»Ε®Π¬-ΉΣΫ«ΫαΙΙΓΘ±ξΚλΒΡΥα–‘ΜυΆ≈“―±Μ÷ΛΟς”κΗΟΙΐ≥ΧΦφ»ίΓΘbΘ§‘ΎΤΏκΡ÷–ΝΫΗωœ©±ϊΜυΗ Α±Υα≤–Μυ÷°ΦδΒΡΜΖΚœΗ¥Ζ÷ΫβΖ¥”Π–Έ≥…ΧΦΜυΝ§Ϋ”Θ§ Ι N ΕΥΚΆ C ΕΥΗϋΫ”ΫϋΘ§¥”Εχ Βœ÷”––ßΒΡ¥σΜΖΡΎθΘΑΖΜ·ΓΘΥφΚσ«–Εœœ©ΧΰΝ§Ϋ”Θ§≤ΔΆ®ΙΐΦΗ¥ΈΫΜ≤φΗ¥Ζ÷ΫβΖ¥”ΠΘ§Ω…ΜώΒΟάύΥΤ”ΎΧλ»Μ≤ζΈο¬μΙΰΖ®ΈςΝ÷ B ΒΡκΡ¥σΜΖΓΘ
‘Ύ’β“ΜΩΣ¥¥–‘ΙΛΉς÷°ΚσΘ§ΤδΥϊΦΗΗω―–ΨΩ–ΓΉι“≤Ά®ΙΐΜυ”Ύœ©ΧΰΒΡΓΑ÷ßΦήΓ±≥…ΙΠΈ»Ε®ΝΥκΡ÷–ΒΡΕΰΦΕΫαΙΙ‘ΣΦΰΘ§≤ΜΙΐ’β–©Η≈Ρν≤ΔΖ«±ΨΉέ ωΒΡ÷ΊΒψΘ§ΕΝ’ΏΩ…≤ΈΩΦΤδΥϊΈΡœΉΓΘ¬ό±ω―ΖΦΑΤδΆ§ ¬ΖΔœ÷ΝΥΟ·Ϋπ τ¥ΏΜ·ΒΡΗ¥Ζ÷ΫβΖ¥”Π‘Ύ¥ΌΫχκΡ¥σΜΖΜ·ΖΫΟφΒΡ≤ΜΆ§”Π”ΟΓΘΥϊΟ«Κ§œ©ΧΰΒΡΝ§Ϋ”Χε”…ΝΫΗω≤Ώ¬‘–‘Ζ≈÷ΟΒΡœ©±ϊΜυΗ Α±Υα≤–Μυ÷°ΦδΒΡΫΜ≤φΗ¥Ζ÷ΫβΖ¥”Π–Έ≥…Θ§ ΙκΡΒΡ C ΕΥΚΆ N ΕΥΗϋΩΩΫϋΘ§¥”ΕχΆ®Ιΐ≥ΘΙφΒΡΡΎθΘΑΖΜ·Μ·―ߥΌΫχ¥σΜΖΜ·Θ®ΆΦ 7bΘ©ΓΘΗΟΖΫΖ®±Μ”Π”Ο”ΎάύΥΤ”ΎΧλ»Μ≤ζΈο¬μΙΰΖ®ΈςΝ÷ΒΡΤΏκΡΒΡΆΖΈ≤ΜΖΜ·ΓΘ‘ΎΑ≤ΉΑΝΥΧΦΜΖΝ§Ϋ”ΧεΚσΘ§”κ FDPPΘ®ΈεΖζ±ΫΜυΕΰ±ΫΜυΝΉθΘΘ©Ζ¥”ΠΘ§“‘ 55%ΒΡ≤ζ¬ ΒΟΒΫΜΖΜ·―ή…ζΈοΓΘ
“λ«ηΥαθΞΚΆΕύΉιΖ÷Ζ¥”ΠΒΡ”Π”Ο
ΈΎΦΣΥΡΉιΖ÷Ζ¥”ΠΘ®U-4CRΘ© «ΙΙΫ®Μυ”ΎκΡΥΊΩρΦήΒΡΕύΙΠΡήΙΛΨΏΓΘΈΛΥΙ‘ΦΚΚΦΑΤδΆ§ ¬ΫΪ’β÷÷ΕύΉιΖ÷Ζ¥”Π”Π”Ο”ΎΚ§ RGD ΜΖΉ¥κΡΥΊΒΡΚœ≥…Θ®ΆΦ 8aΘ©ΓΘΥϊΟ«ΒΡ≤Ώ¬‘…φΦΑ»ΐΗωΝ§–χΒΡΈΎΦΣΖ¥”ΠΓΘ’β«ΑΝΫΗωΝ§–χΒΡ Ugi ΥΡΉιΖ÷Ζ¥”ΠΙΙΫ®ΝΥΚ§ RGD ΒΡΖ«ΜΖΉ¥«ΑΧεΓΘΆ―±ΘΜΛΚσΘ§‘Ό≤…”ΟΝμ“ΜΗω Ugi »ΐΉιΖ÷ΥΡ÷––ΡΖ¥”Π Βœ÷κΡΡΘΡβΈοΒΡΜΖΜ·ΓΘ
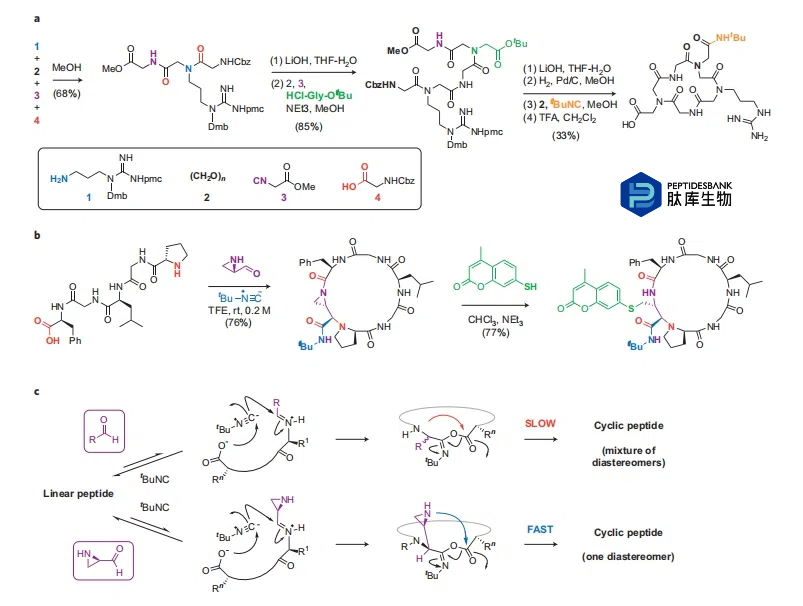
ΆΦ 8 | ΕύΉιΖ÷Ζ¥”ΠΫιΒΦΒΡκΡΜΖ±’ΚœΓΘaΘ§Ά®ΙΐΝ§–χΒΡ U-4CR Ζ¥”ΠΚœ≥…ΜΖΉ¥ RGD ΈεκΡάύΜ·ΚœΈοΓΘ Ήœ»≤…”ΟΝΫΗωΝ§–χΒΡ U-4CR Ζ¥”ΠΙΙΫ®Ζ«ΜΖΉ¥«ΑΧεΘ§ΥφΚσΕ‘ΤδΕΥΜυΫχ––Ά―±ΘΜΛ¥ΠάμΘ§‘ΌΆ®Ιΐ Ugi »ΐΉιΖ÷ΥΡ÷––ΡΖ¥”Π Βœ÷κΡάύΜ·ΚœΈοΒΡΜΖΜ·ΓΘbΘ§Ά®Ιΐ”κΝΫ–‘ΜΖ―θ““Άι»©ΚΆ“λ«ηΜ·ΈοΒΡΕύΉιΖ÷Ζ¥”ΠΫΪΝυκΡœό÷Τ‘Ύ“ΜΗω¥σΜΖ÷–ΓΘΥυΒΟΚ§ N-θΘΜυΜΖ―θ““ΆιΒΡ¥σΜΖΥφΚσΩ…Ά®Ιΐ”κ 7-έœΜυ-4-ΦΉΜυœψΕΙΥΊΒΡΩΣΜΖΖ¥”ΠΫχ––ΈΜΒψΧΊ“λ–‘–ό ΈΓΘcΘ§Ugi ΫιΒΦΒΡ”κΜΖ―θ““Άι»©ΒΡΜΖΜ·Ζ¥”Π÷–Ιέ≤λΒΫΒΡΗΏΖ«Ε‘”≥―Γ‘ώ–‘ΒΡΩ…ΡήΫβ ΆΓΘ
Νμ“ΜΗω Ι”Ο U-4CR ΒΡάΐΉ”ά¥Ή‘ Kazmaier ΦΑΤδΆ§ ¬ΒΡ Β―ι “ΓΘΥϊΟ«ΙΙΫ®ΜΖΉ¥κΡΡΘΡβΈοΒΡΖΫΖ®…φΦΑ N-ΆιΜυΜ·Α±ΜυΥαΘ§’β–©Α±ΜυΥα±Μ’ϊΚœΒΫΥΡΉιΖ÷ Ugi Ζ¥”Π÷–“‘ΙΙΫ®œΏ–‘«ΑΧεΘ§ΥφΚσΫχ––ΜΖΜ·Η¥Ζ÷ΫβΖ¥”ΠΘ®RCMΘ©ΓΘΆ®Ιΐ’β÷÷ΖΫΖ®Θ§Ω…“‘‘ΎΜΖ÷–ΒΡ»ΈΚΈΈΜ÷ΟΖ≈÷Ο≤ΜΆ§ΒΡΦΪ–‘ΓΔ«ΉΥ°–‘ΚΆ ηΥ°–‘ΜυΆ≈ΓΘ
ΒΛΡα…αΖρΥΙΜυΦΑΤδΆ§ ¬ΩΣΖΔΝΥ“ΜœΒΝ––¬”±ΒΡ“λ«ηΥαθΞΫιΒΦΒΡθΘΑΖ≈ΦΝΣΖΫΖ®Θ§”Ο”ΎΚœ≥…ΨΏ”–Χτ’Ϋ–‘ΒΡ N,N-ΕΰΆιΜυΜ·θΘΑΖΓΘ‘ΎΉνΫϋΒΡ“ΜœνΫή≥ωΙΛΉς÷–Θ§ΥϊΟ«ΫΪΗΟΖΫΖ®”Π”Ο”ΎΜΖφΏΥΊ A ΒΡ»ΪΚœ≥…ΓΘΆ®Ιΐ“ΜœΒΝ–“λ«ηΥαθΞΫιΒΦΒΡ≈ΦΝΣΖ¥”ΠΫΪΗςΗωκΡΒΞ‘ΣΝ§Ϋ”‘Ύ“ΜΤπΓΘ
Ψ≤ΒγΩΊ÷ΤΒΡΜΖΜ·Ζ¥”Π
ΕΧκΡ‘ΎΥ°»ή“Κ÷–Ά®≥Θ≥ ΈόΙφΨμ«ζΉ¥Χ§Θ§’β «”…”ΎΥ°ΡήΙΜΤΤΜΒκΡΝ¥ΡΎΒΡ«βΦϋΓΘ»ΜΕχΘ§‘ΎΦΪ–‘”–Μζ»ήΦΝ÷–Θ§ΕΧœΏ–‘κΡ«ψœρ”Ύ–Έ≥…ΜΖΉ¥ΙΙœσΘ§’β «”… N ΕΥΚΆ C ΕΥ÷°ΦδΒΡάκΉ”Ε‘Ής”ΟΥυ«ΐΕ·ΒΡΓΘάϊ”Ο’β÷÷––ΈΣΩ…ΡήΜαΩΣΖΔ≥ω”––ßΒΡΜΖΜ·≤Ώ¬‘Θ§“ρΈΣ÷Φ‘Ύ‘Ύ’ϊΗωΜΖΜ·Ιΐ≥Χ÷–Έ§≥÷ C ΕΥΚΆ N ΕΥ÷°ΦδάκΉ”Ε‘Ής”ΟΒΡΖ¥”ΠΡήΙΜάϊ”ΟΨ≤Βγ”’ΒΦΒΡ‘ΛΜΖΜ·ΙΙœσΧεΓΘΈΣ¥ΥΘ§”»ΕΓΦΑΤδΆ§ ¬Ε‘Έ¥±ΘΜΛΒΡ NH ≈ΦΒΣΜΖ»©ΓΔ“λ«ηΥαθΞΚΆœΏ–‘κΡΫχ––ΝΥΖ¥”ΠΘ®ΆΦ 8bΘ©ΓΘ
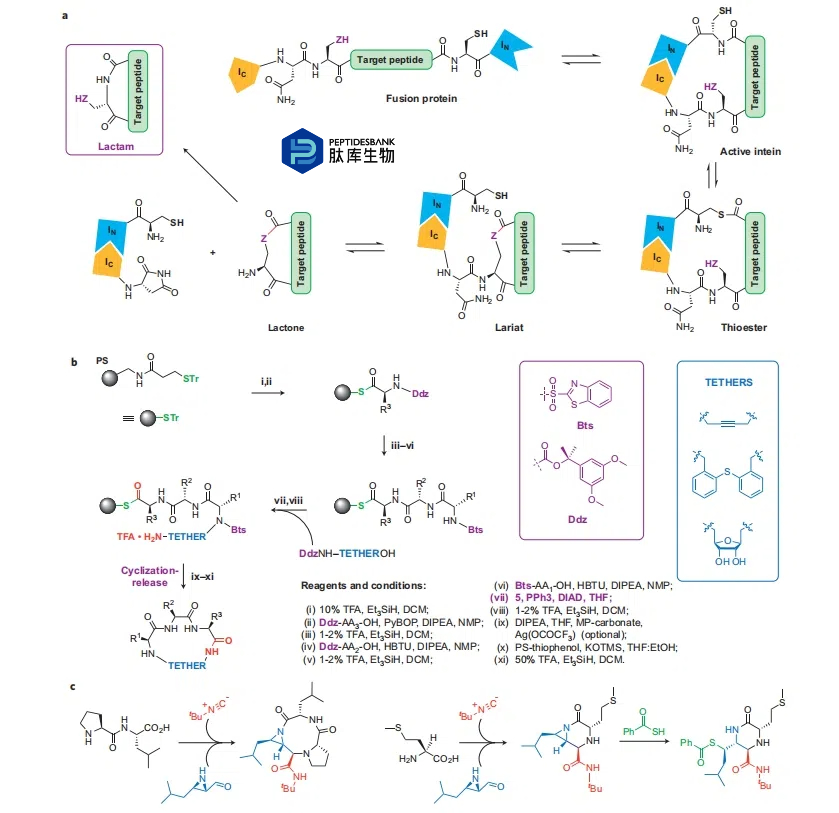
ΆΦ 9 | κΡ¥σΜΖΜ·ΚœΈοΩβΩΣΖΔ≤Ώ¬‘ΓΘaΘ§”Ο”ΎΚœ≥…ΜΖκΡΒΡΖ÷Ν―ΡΎκΡΟΗΫιΒΦΒΡΜΖΜ·Ν§Ϋ”ΓΘbΘ§”Ο”ΎΩΣΖΔΜΖκΡΡΘΡβΈοΩβΒΡ MATCH ΦΦ θΖ¥”ΠΖΫΑΗΓΘ’Ι ΨΝΥΥυ”ΟΗς÷÷Ν§Ϋ”ΧεΒΡΦΗΗω ΨάΐΘ®“‘άΕ…Ϊœ‘ ΨΘ©ΓΘcΘ§ ΐΉ÷ΈΔΝςΩΊΦΦ θ“―”Ο”ΎΆ®ΙΐΉιΚœ ‘ΦΝΒΡάκ…Δ“ΚΒΈά¥¥¥Ϋ®ΜΖκΡΩβΓΘ’β Βάΐ±μΟςΘ§ΕΰκΡ H-Pro-Leu-OH ΚΆΑ±ΜυΥαΦΉΝρΑ±Υα‘ΎΆ§“ΜΗω–ΨΤ§Ζ¥”ΠΤς÷–Ά®Ιΐ”κΒΣ±ϊύΛ»©ΚΆ“λ«ηΜ·ΈοΒΡ“ΚΒΈΜλΚœΕχΆ§ ±ΜΖΜ·ΓΘΥφΚσΘ§Ά®ΙΐΫχ“Μ≤Ϋ”κΚ§Νρ±ΫΦΉΥαΒΡ“ΚΒΈΜλΚœΘ§ Βœ÷ΝΥΦΉΝρΑ±Υα≤ζΈοΒΡΈΜΒψΧΊ“λ–‘–ό ΈΓΘDCMΘ§Ε଻ֹΆιΘΜDIADΘ§Εΰ“λ±ϊΜυ≈ΦΒΣΕΰΦΉΥαθΞΘΜHBTUΘ§2-(1H-±Ϋ≤Δ»ΐΏρ-1-Μυ)-1Θ§1Θ§3Θ§3-ΥΡΦΉΜυκεΝυΖζΝΉΥαθΞΘΜKOTMSΘ§»ΐΦΉΜυΙηΦΊΓΘ
’β÷÷ΕύΉιΖ÷ΖΫΖ®ΡήΗΏ–ßΒΊ…ζ≥…ΨΏ”–ΗΏΜ·―ß―Γ‘ώ–‘ΚΆΝΔΧε―Γ‘ώ–‘ΒΡΜΖκΡΘ§«“Έό“λΙΙΜ·ΓΔΜΖΕΰΨέΜ·ΜρΒΆΨέΜ·ΒΡΦΘœσΓΘΨΏ”–Χτ’Ϋ–‘ΒΡ÷–Β»¥σ–ΓΜΖκΡ“Ή”Ύ÷Τ±ΗΘΜΖ¥”Π ±Φδ≤ΜΒΫ 10 –Γ ±Θ§≤ζ¬ ΗΏΓΘΗΟΖ¥”ΠΡή“‘ΗΏΡΠΕϊ≈®Ε»…ζ≥…κΡ¥σΜΖΘ§”κœΏ–‘κΡ«ΑΧεΒΡ≥ΛΕ»ΈόΙΊΓΘΨί»œΈΣΘ§ΜΖ±ϊΆι»©ΒΡ»©Μυ÷––Ρ”κœΏ–‘κΡ«ΑΧεΒΡ N ΕΥ÷°Φδ–Έ≥…ΒΡ―«ΑΖάκΉ” «”…Ψ≤ΒγΈ»Ε®ΒΡάκΉ”Ε‘ΧαΙ©ΒΡΓΘ”»ΕΓΦΑΤδΆ§ ¬Χα≥ωΘ§ΝΫ–‘ΜΖ±ϊΆι»©ΒΡΠΝΈΜ¥φ‘Ύ«ΉΚΥ÷––Ρ «ΗΏ≤ζ¬ ΚΆΗΏΖ«Ε‘”≥―Γ‘ώ–‘ΒΡ‘≠“ρΓΘΒ±‘ΎΖ¥”Π÷– Ι”ΟΒΞΙΌΡήΆ≈»©”κ“λ«ηΥαθΞΚΆκΡ ±Θ§Ιέ≤λΒΫΒΆΖ«Ε‘”≥―Γ‘ώ–‘Θ®ΆΦ 8cΘ©ΓΘΗϋ÷Ί“ΣΒΡ «Θ§Ά®≥Θ‘ΎœΏ–‘κΡΘ®…Ό”ΎΝυΗω≤–ΜυΘ©ΒΡ U-4CR ΜΖΜ·Ιΐ≥Χ÷–ΖΔ…ζΒΡ≤ΜΤΎΆϊΒΡΜΖΕΰΨέΜ·œ÷œσΈ¥±ΜΙέ≤λΒΫΓΘΜΖκΡΗΟ≤ζΤΖΑϋΚ§“ΜΗωΒΣ±ϊύΛ–ό ΈΈΜΒψΘ§‘ΎΚœ≥…ΒΡΚσΤΎΫΉΕΈΩ…“‘‘Ύ¥ΥΈΜΒψΝ§Ϋ”κΡάύΚΆΖ«κΡάύ≤ύΝ¥ΓΘ
Ϋα¬έ”κ’ΙΆϊ
ΚσΜυ“ρΉι ±¥ζΫΪΦΧ–χΧαΙ©”–ΙΊ…ζΈοΑ–ΒψΒΡ–¬–≈œΔΘ§”»Τδ «ΒΑΑΉ÷ - ΒΑΑΉ÷ œύΜΞΉς”ΟΖΫΟφΒΡΓΘ’βάύΡ―“‘ΙΞΩΥΒΡΈ ΧβΈόΖ®”Ο–ΓΖ÷Ή”ά¥ΫβΨωΓΘΥφΉ≈‘Ϋά¥‘ΫΕύΒΡΒΑΑΉ÷ - ΒΑΑΉ÷ œύΜΞΉς”Ο±Μ÷Λ ΒΈΣ÷ΈΝΤΗ…‘ΛΒΡΡΩ±ξΘ§Ε‘ΡήΙΜΫχ––≥Λ ±ΦδœύΜΞΉς”ΟΒΡΖ÷Ή”ΒΡ–η«σΫΪΜα≤ΜΕœ‘ω≥ΛΓΘ’κΕ‘¥ΥάύΖ÷Ή”ΒΡΝιΜνΚœ≥…≤Ώ¬‘”ΠΒ±ΡήΙΜ Βœ÷ΚσΤΎΙΙœσΒς’ϊΚΆ»ήΫβΕ»”≈Μ·ΓΘΉν÷’ΡΩ±ξ «ΒςΫΎΡήΙΜ¥©ΙΐœΗΑϊΡΛΒΡΖ÷Ή”÷–ΒΡΕΰΦΕΫαΙΙΓΘΥφΉ≈Κœ≥…ΖΫΖ®ΒΡΗ¥‘”–‘≤ΜΕœΧαΗΏΘ§Έ“Ο«ΩœΕ®ΜαΦϊ÷Λ–¬–ΆΟς»Ζ±μΟφΒΡΖΔœ÷Θ§Έ“Ο«ΫΪΤδ≥ΤΈΣΧΊ»®’έΒΰΓΘκΡ¥σΜΖΈΣΧΫΨΩ¥ΥάύΗ¥‘”ΫαΙΙΧαΙ©ΝΥάμœκΒΡΜζΜαΓΘ
≤ΈΩΦΈΡœΉΘΚDOI: 10.1038/NCHEM.1062
≤ΈΩΦΈΡœΉΘΚ±ΨΈΡΈΣ––“ΒΫΜΝς―ßœΑΘ§Αφ»®Ιι‘≠Ής’ΏΥυ”–Θ§»γ”–«÷»®Θ§Ω…ΝΣœΒ…Ψ≥ΐ
