
’Σ“Σ:
ΒΑΑΉ−ΒΑΑΉœύΜΞΉς”Ο (PPIs) ‘ΎΒςΫΎΜζΧε…ζΟϋΜνΕ·÷–ΤπΉ≈ΨωΕ®–‘ΒΡΉς”Ο, «ΧεΡΎ÷ΎΕύ–≈Κ≈¥ΪΒΦΆ®¬ΖΒΡΙΊΦϋΜζ÷Τ, Τδ÷–ΚήΕύΙΊΦϋΒΑΑΉ÷ «Ω…“‘άϊ”ΟΒΡ«±‘Ύ“©ΈοΑ–ΒψΓΘΧΫΥς”––ßΒςΩΊΒΑΑΉ−ΒΑΑΉœύΜΞΉς”ΟΒΡΖΫΖ®, ΫΪΕ‘…ζάμ―ß“‘ΦΑ“©Έο―–ΨΩ¥σ”–ώ‘“φΓΘΨχ¥σΕύ ΐΒΑΑΉ−ΒΑΑΉœύΜΞΉς”Ο“‘œύΕ‘Ϋœ¥σ«“Ϋœ«≥ΒΡΉς”ΟΟφΡΘ ΫΫχ––, –ΓΖ÷Ή”Ρ―“‘–Έ≥…”––ßΫαΚœΜρΒςΩΊΓΘΫΪΒΑΑΉ−ΒΑΑΉœύΜΞΉς”Ο÷–“‘ΕύκΡΕΰΦΕΫαΙΙΈΣ÷ß≥≈Ι«ΦήΒΡ’έΒΰ―«ΫαΙΙ”ρ÷–ΒΡ ΠΝ-¬ί–ΐκΡΒΞΕάΧα»Γ≥ωά¥, Ά®ΙΐΜ·―ßΚœ≥…ΒΡΖΫΖ®Φ”“‘ΙΙΫ®ΫΪ”–Ω…ΡήΒΟΒΫ―Γ‘ώ–‘Ής”Ο”ΎΑ–±ξΒΑΑΉΒΡΜν–‘ΕύκΡ“©Έοœ»ΒΦΈοΓΘ»ΜΕχ¥σ≤ΩΖ÷ΕύκΡΤ§ΕΈ‘ΎάκΩΣΒΑΑΉ÷ ’ϊΧεΫαΙΙΚσΫΪΈόΖ®Έ»Ε®–Έ≥…ΫαΚœΥυ–ηΒΡΕΰΦΕΫαΙΙ, Εχ“Ή”Ύ–Έ≥…ΈόΙφ‘ρΨμ«ζΙΙœσ¥”ΕχΒΦ÷¬ΫαΚœΜν–‘œ¬ΫΒ, ≤Δ«“Ηϋ“Ή ήκΡΟΗΒΡΫΒΫβ, ΈόΖ®÷±Ϋ”≥…“©ΓΘ”Π”Ο»ΪΧΦΙ«Φή–Έ≥…≤ύΝ¥ΜΖΚœΫαΙΙΗΡ‘λΕύκΡά¥Έ»Ε® ΠΝ-¬ί–ΐκΡΒΡΜν–‘ΙΙœσ, Φ¥Ε© ικΡ (stapled peptide), ≥…ΈΣΩΥΖΰ’β“Μ»±œίΒΡΉν÷±Ϋ”Ήν”––ßΖΫΖ®ΓΘΗΟΖΫΖ®≤ΜΫωΩ…“‘ΧαΗΏΤδ‘≠±ΨΒΡΒΑΑΉΫαΚœΜν–‘, Εχ«“ΨΏ”–ΫœΗΏΒΡ¥ζ–ΜΈ»Ε®–‘ΚΆœΗΑϊΡΛΆ®ΆΗ–‘ΓΘΜυ”Ύ’β–©œ‘÷χΒΡ”≈ Τ, Ε© ικΡ“―Ψ≠≥…ΈΣ“Μάύ÷Ί“ΣΒΡΜν–‘ΕύκΡΫαΙΙΗΡ‘λΖΫ Ϋ, “≤±ΊΫΪ”…¥Υ–Έ≥…ΗϋΕύΒΡ“‘ΒΑΑΉ−ΒΑΑΉœύΜΞΉς”ΟΈΣΑ–ΒψΒΡ–¬–ΆΕύκΡ“©ΈοΓΘ±ΨΈΡΫΪΉ≈÷ΊΉέ ω≤ΔΧ÷¬έΆ®ΙΐΜ·―ß ÷ΕΈΚœ≥…Ε© ικΡΒΡΖΫΖ®ΦΑΤδ“©άμΜν–‘―–ΨΩΫχ’ΙΓΘ
ΒΑΑΉ−ΒΑΑΉœύΜΞΉς”Ο (PPIs) «ΧεΡΎ÷ΎΕύ–≈Κ≈¥ΪΒΦΆ®¬ΖΒΡΙΊΦϋΜζ÷Τ÷°“Μ, “≤ «ΫϋΡξά¥“©Έο―–ΨΩΒΡ»»Βψ, ”–―–ΨΩΙάΥψ»ΥΧεΡΎΉ‘…μΧεœΗΑϊ÷–”–”κΦ≤≤ΓœύΙΊΒΡ PPI Ής”Ο‘Ύ 13 ΆρΒΫ 65 ΆρΗω÷°Φδ[1], ’βΈό“…ΈΣ―–ΖΔ÷ΈΝΤ÷ν»γΑ©÷ΔΓΔΟβ“ΏΦ≤≤ΓΓΔ―Ή÷ΔΓΔΗ–»ΨΒ»Φ≤≤ΓΒΡ–¬–Ά“©ΈοΧαΙ©ΝΥ“ΜΗωΨό¥σΒΡΑ–ΒψΩβΓΘΆ®ΙΐΕ‘ΒΑΑΉΗ¥ΚœΈοΒΡΫαΙΙ―–ΨΩ±μΟς, ¥σ≤ΩΖ÷ PPI «“‘ΒΑΑΉ÷ ΦδΕύΗωΕύκΡΕΰΦΕΫαΙΙΒΞ‘Σ“‘“Μ÷÷ΕύΈΜΒψΒΡ¥σΟφΜΐΫαΚœΖΫ Ϋ (ΤΫΨυΉς”ΟΟφΜΐΈΣ 1 500ΓΪ3 000 Å2) ΖΔ…ζΫαΚœ[2], ΆυΆυ≤ΜΨΏ”–ΧΊΕ®ΒΡΫαΚœΩΎ¥ϋΓΘ’βœ‘÷χ‘ωΦ”ΝΥ“‘ PPI ΈΣΑ–ΒψΩΣΖΔ≥ΘΙφΒΡ–ΓΖ÷Ή”“©ΈοΡ―Ε»[3]ΓΘΥφΉ≈ΒΞΩΥ¬ΓΩΙΧεΦΦ θΒΡΖΔ’ΙΕχ≤ζ…ζΒΡ÷ΈΝΤ–‘ΒΞΩΙ“©Έο”–ΡήΝΠ“‘ PPI ΈΣΑ–Βψ, ≤Δ“―‘ΎΩΙ÷ΉΝωΓΔΟβ“ΏΒςΫΎΓΔΩΙΗ–»ΨΒ»ΖΫΟφ»ΓΒΟΝΥΨό¥σΒΡ≥…ΙΠΓΘΒΪΡΩ«ΑΒΞΩΙ“©Έο≥…±ΨΗΏΓΔΦΦ θΡ―Ε»¥σΓΔ“©ΤΖΦέΗώΗΏΒΡΧΊΒψ, ΦΪ¥σΒΡœό÷ΤΝΥΤδΙψΖΚ”Π”Ο; ¥ΥΆβΒΞΩΙΉςΈΣ¥σΖ÷Ή”ΒΑΑΉΚήΡ―Ϋχ»κœΗΑϊ, ΚήΕύΦ≤≤ΓœύΙΊΒΡœΗΑϊΡΎΒΑΑΉΒΡΒςΩΊΩΩΒΞΩΙ“©ΈοΡ―“‘ Βœ÷[4, 5]ΓΘ
‘ΎΒΑΑΉ÷ ÷–, ΠΝ-¬ί–ΐ «ΉνΈΣΤ’±ιΒΡΕΰΦΕΫαΙΙΒΞΈΜ, ≤Δ«“‘ΎΒΑΑΉ−ΒΑΑΉΓΔΒΑΑΉ-DNA ΒΡœύΜΞΉς”Ο÷–ΖΔΜ”ΦΪΤδ÷Ί“ΣΒΡΉς”ΟΓΘΚήΕύΒΑΑΉ−ΒΑΑΉœύΜΞΉς”ΟΆ®Ιΐ“‘ΕΰΦΕΫαΙΙΈΣ÷ß≥≈Ι«ΦήΒΡ’έΒΰ―«ΫαΙΙ”ρ Βœ÷, Εχ ΠΝ-¬ί–ΐΕΰΦΕΫαΙΙΒΡκΡΝ¥ «Τδ÷–ΉνΈΣΤ’±ιΒΡΉς”ΟΤ§ΕΈ[6]ΓΘ’β–© ΠΝ-¬ί–ΐκΡΒΡΤΫΨυ≥ΛΕ»ΫœΕΧ, “ρ¥ΥΒΞΕάΙΙΫ®’βάύ ΠΝ-¬ί–ΐκΡ≤ΔΆ®ΙΐΜ·―ßΚœ≥…ΒΟΒΫΩ…”κΑ–±ξΒΑΑΉΖΔ…ζ―Γ‘ώ–‘Ής”ΟΒΡΜν–‘ΕύκΡ“©Έο «Ω…––ΒΡΓΘΠΝ-¬ί–ΐ «Α±ΜυΥαΆ®ΙΐκΡΦϋ–Έ≥…ΒΡΨΏ”–”“ ÷¬ί–ΐΕΰΦΕΫαΙΙΒΡΕύκΡ, ¬ί–ΐΟΩ÷ήΑϋΚ§ 3.6 ΗωΑ±ΜυΥα≤–Μυ, ¬ίΨύΈΣ 0.54 nmΓΘΗΟΫαΙΙ”…ΒΎi ΗωΑ±ΜυΥακΡΦϋΒΡ N-H ”κΒΎ i+4 ΗωΑ±ΜυΥακΡΦϋΒΡτ Μυ―θ–Έ≥…«βΦϋΕχΙΙ≥…ΒΡ °»ΐ‘ΣΓΑΜΖΓ±ΥυΈ»Ε®[7]ΓΘ”…”ΎκΡΝ¥Ι«Φή“‘¬ί–ΐΒΡΖΫ Ϋ»Τ÷α…λ’Ι, Τδ÷ςΝ¥Ή‘”…–ΐΉΣ±Μœό÷Τ, ΒΪΕύκΡ≤ύΝ¥Ω…“‘‘ΎΆβ≤Ω…λ’Ι¥”Εχ±μœ÷≥ωΕάΧΊΒΡΆΊΤΥΫαΙΙ, ’β―υΒΡΧΊ–‘ ΙΒΟ ΠΝ-¬ί–ΐΕύκΡΩ…“‘‘Ύ PPIΙΐ≥Χ÷–ΨΪ»ΖΤθΚœΥυΕ‘”ΠΒΡ»ΐΈ§ΫαΙΙ”ρΓΘΆ≥ΦΤ±μΟς≤Έ”κPPIs ΒΡ ΠΝ-¬ί–ΐΤ§ΕΈΤΫΨυ≥ΛΕ»ΫœΕΧ, ΤΫΨυΈΣ 2 ÷Ν 3 Ηω¬ί–ΐ≥ΛΕ» (Μρ 8ΓΪ12 ΗωΑ±ΜυΥα≤–Μυ)[8]ΓΘ»ΜΕχ»γΙϊ¥”ΒΑΑΉ÷ ΫαΙΙ÷–ΫΊ»ΓΗΟ ΠΝ-¬ί–ΐΤ§ΕΈ, ÷±Ϋ”Κœ≥…ΒΡΕΧκΡΤ§ΕΈ‘ΎΥ°»ή“Κ÷–≤ΜΡή±Θ≥÷Έ»Ε®ΒΡ ΠΝ-¬ί–ΐΕΰΦΕΫαΙΙ≤Δ«ψœρ”Ύ–Έ≥…ΈόΙφ‘ρΨμ«ζ[9]ΓΘΈόΙφ‘ρΨμ«ζΙΙœσΒΡΕύκΡ‘Ύ”κΑ–±ξΫαΚœ ±–ηΉΣ±δΈΣ ΠΝ-¬ί–ΐΙΙœσ, ΗυΨίΫαΚœΉ‘”…ΡήΦΤΥψΙΪ Ϋ:
Τδ÷–λΊ±δ(ΠΛS=ΠΛSW+ΠΛSvib−ΠΛSrt−ΠΛSint,DSWΈΣΥ°λΊ,ΠΛSvibΈΣ’ώΕ·λΊ,ΠΛSrtΈΣ≈δΧεΒΡΤΫΕ·ΉΣΕ·λΊ,ΠΛSintΈΣ≈δΧεΒΡΒΞΦϋ–ΐΉΣλΊ)ΓΘΕ‘”ΎΕύκΡ’β÷÷÷ςΝ¥ΈΣ»α–‘ΫαΙΙΒΡ¥σΖ÷Ή”Μ·ΚœΈοΕχ―‘,‘Ύ”κ ήΧεΫαΚœ«ΑΚσΒΡΒΞΦϋ–ΐΉΣλΊΠΛSintΚΆΤΫΕ·ΉΣΕ·λΊΠΛSrtΒΡ±δΜ· «Ψό¥σΒΡ,Υυ“‘»γΙϊΜν–‘ΕύκΡ≤ΜΡή±Θ≥÷Τδ‘Ύ”κΑ–±ξΒΡΫαΚœ«ΑΚσΒΡΙΙœσ“‘ΦΑΖ÷Ή”ΡΎ«βΦϋΒΡΈ»Ε®,ΤδΫαΚœΙΐ≥ΧΫΪ“ΐΤπΚή¥σΒΡλΊΥπ ß(ΠΛSΗΚ÷Β‘ω¥σ),¥”ΕχΒΦ÷¬ΫαΚœΡήΠΛGœ¬ΫΒΖυΕ»±δ–Γ,…ζΈοΜν–‘ΫΒΒΆΓΘ¥ΥΆβ’β÷÷ φ’ΙΒΡΙΙœσ≥δΖ÷±©¬ΕκΡΒΡΟΗΫβΈΜΒψ, ‘ωΦ”ΝΥκΡΟΗΒΡΥ°ΫβΦΗ¬ , ΒΦ÷¬ΕύκΡΦΪ“Ή±ΜΫΒΫβ, …ζΈοΈ»Ε®–‘ΒΆ, «“≤Μ“Ή”Ύ¥©ΙΐœΗΑϊΡΛ[10]ΓΘ
“ρ¥Υ, ΧΫΥςΗς÷÷”––ßΒΡΖΫ ΫΈ»Ε®ΕύκΡΒΡ ΠΝ-¬ί–ΐΙΙœσ, Μρ’Ώ“‘Ζ«Χλ»ΜΒΡΙ«ΦήΫαΙΙΙΙΫ® ΠΝ-¬ί–ΐκΡΒΡΡΘΡβΈο≥…ΈΣΜ·―ß…ζΈο―ß“‘ΦΑΕύκΡ“©ΈοΜ·―ß―–ΨΩΒΡ»»Βψ[11]ΓΘΗυΨί ΠΝ-¬ί–ΐΒΡΧΊΒψ, κΡΝ¥ i, i+4, i+7, i+11 ΈΜΒψ…œΒΡ≤ύΝ¥¥Π”Ύ¬ί–ΐΫαΙΙΆ§≤ύ, ‘Ύ i ΚΆ i+4 Μρ i ΚΆ i+7 ÷°ΦδΒΡ≤ύΝ¥ΜυΆ≈…œΆ®ΙΐΫΜ≤φ≈ΦΝΣ–Έ≥…Ν§Ϋ”«≈ά¥Έ»Ε® ΠΝ-¬ί–ΐΒΡΙΙœσ «“Μ÷÷––÷°”––ßΒΡΖΫΖ®, ΗΟΖΫΖ®”…”ΎΝ§Ϋ”«≈άύΥΤΕ© ιΕΛΫΪ ΠΝ-¬ί–ΐΫαΙΙΦ”“‘ΙΧΕ®, Ι ≥Τ÷°ΈΣΕ© ικΡ (stapled peptide)ΓΘVerdine Β―ι “±®ΒάΝΥΫΪ BCL-2 ΒΑΑΉ…ηΦΤΗΡ‘λΈΣΨΏ”–Ε© ικΡΫαΙΙΒΡΜν–‘Τ§ΕΈΒΡ―–ΨΩ, ΗΡ‘λΚσΒΡBCL-2Ε© ικΡΤ§ΕΈΡή≤Έ”κœΗΑϊΡΎΒΡ PPI≤Δ«“±μœ÷≥ωΝΥ“ΜΕ®ΒΡΧεΡΎ…ζΈο―ßΜν–‘[12], “ΜΨΌ¥ρΤΤΝΥ“Μ–©ΩΤ―ßΦ“Χα≥ωΒΡΓΑΖ«≥…“©–‘ΒΑΑΉ (undruggable proteins)Γ±ΒΡΥΦΈ§ΫϊοάΓΘΥφΚσΙζΡΎΆβΒΡΩΈΧβΉι“≤ΕΦ¬Ϋ–χΩΣ’ΙΝΥΕ© ικΡ‘ΎΑ©÷ΔΓΔΑ§ΉΧ≤Γ“‘ΦΑœΗΑϊ–≈Κ≈Ά®¬ΖΖΫΟφΒΡ―–ΨΩ, ”…¥Υ Ω…Φϊ, Ε© ικΡ’β“Μ–¬–ΆΕύκΡΫαΙΙΗΡ‘λΖΫΖ®ΫΪΕ‘ΕύκΡ“©ΈοΒΡ―–ΖΔ“‘ΦΑœύΙΊΜυ¥Γάμ¬έΒΡ―–ΨΩΧαΙ©“ΜΧθ–¬”±ΓΔ±ψΫίΓΔΩ…––ΒΡΫβΨωΖΫΑΗΓΘΈΣ¥Υ, ±ΨΈΡΉέ ωΝΥΗΟΝλ”ρΫϋΡξά¥±®ΒάΒΡΕ© ικΡΗΡ‘λΒΡΖΫΖ®ΓΔ≥…ΙΠ Βάΐ“‘ΦΑœύΙΊ―–ΨΩΫχ’Ι, “‘ΤΎΈΣΗΟΝλ”ρΒΡ―–ΨΩΧαΙ©≤ΈΩΦΚΆΫηΦχΓΘ
ΡΩ«ΑΕ© ικΡΑ¥’’«≈Ν§ΫαΙΙ (Ε© ι«≈) ΒΡάύ–Ά≤ΜΆ§Ω…Ζ÷ΈΣΝΫ¥σάύ, Τδ“ΜΈΣ“‘ΧΦ−ΧΦ≈ΦΝΣΖ¥”Π–Έ≥…ΒΡ»ΪΧΦΝ¥Ε© ι«≈Ε© ικΡ, Νμ“ΜάύΈΣ“‘‘”‘≠Ή”ΉςΈΣΝ§Ϋ”Ζ¥”Π÷––ΡΒΡΚ§‘”‘≠Ή”Ν¥Ε© ι«≈Ε© ικΡ, œ¬ΟφΫΪΕ‘ΗΟΝΫ’ΏΖ÷±πΫχ––Ϋι…ήΓΘ
1 »ΪΧΦ«βΝ¥Ε© ικΡΒΡΚœ≥…ΦΑΜν–‘ΗΡΙΙΈο
1.1 »ΪΧΦΝ¥Ε© ικΡΒΡΚœ≥…ΦΑΫαΙΙ“ΣΥΊ
ΧΦΝ¥Ε© ι«≈ΧΊ±π «œ©ΜυΕ© ι«≈ «Κœ≥…Ε© ικΡΒΡΨ≠ΒδΖΫΖ®, 1994 Ρξ”… Grubbs –ΓΉι±®ΒάΝΥάϊ”ΟΤδΖΔœ÷ΒΡœ©ΧΰΙΊΜΖΗ¥Ζ÷ΫβΖ¥”Π (RCM Ζ¥”Π) Ϋχ––ΕύκΡΒΡΫαΙΙΗΡ‘λ[13], ΗΟΖΫΖ®÷ς“Σ‘ΎΚœ≥…ΕύκΡΚσΆ®ΙΐΕ‘κΡΝ¥ i ΚΆi+4 ΈΜΒΡΥΩΑ±ΥαΫχ––≤ύΝ¥œ©±ϊΜυΟ―Μ·―ή…ζ‘ΌΆ®ΙΐRCM Ζ¥”ΠΒΟΒΫ”…ΜΖœ©Μυ«≈Έ»Ε®ΒΡa-¬ί–ΐκΡ (ΆΦ 1A)ΓΘVerdine ΦΑΤδΆ≈Ε”‘Ύ¥Υ÷°ΚσΖΔ’Ι≥ωάϊ”ΟΨΏ”–Ρ©ΕΥœ©Χΰ≤ύΝ¥ΒΡΖ«Χλ»ΜΑ±ΜυΥαΧφΜΜΧλ»ΜΕύκΡ÷–ΒΡΑ±ΜυΥα≤–Μυ≤ΔΆ®Ιΐ RCM Ζ¥”Π–Έ≥…ΜΖœ©«≈ (ΆΦ 1B)ΓΘΗΟΖΫΖ®ΫβΨωΝΥ Grubbs ΖΫΖ®÷–”…”Ύ‘ΎΚœ≥…ΕύκΡΚσ‘ΌΫχ––œ©±ϊΜυ»Γ¥ζΥυ¥χά¥ΒΡΕ‘”ΎΕύκΡ≤ύΝ¥ΫαΙΙΒΡœό÷Τ“Σ«σ“‘ΦΑ“ΚœύΜΖΚœΚœ≥…ΒΡ≤Μ±ψ, ≤Δ Ή¥ΈΧα≥ωΝΥΓΑΕ© ικΡΗΡ‘λ(peptide stapling)Γ±ΒΡΗ≈Ρν[11], ≤ΔΨ≠Ιΐ≤ΜΕœΒΡΖΔ’ΙΚΆΗΡΫχ≥…ΈΣΝΥ“Μ÷÷Ψ≠ΒδΒΡΤ’ –‘ΕύκΡΗΡ‘λ≤Ώ¬‘ΚΆΖΫΖ®ΓΘ

1.1.1 ΜΖœ©Ε© ικΡΦΑΫαΙΙ“ΣΥΊ »γ«ΑΥυ ωΒΞΕ© ι«≈
ΜΖœ©Ε© ικΡ «“Μ÷÷Ψ≠ΒδΒΡΕ© ικΡ–Έ Ϋ, “≤ «Ε‘”ΎΕ© ικΡΒΡΚœ≥…ΓΔΫαΙΙ“‘ΦΑΜν–‘±®ΒάΉνΕύΒΡ―–ΨΩΖΫœρΓΘΡΩ«ΑΫœΈΣ≥… λΒΡΖΫΖ® «“‘ν…¥ΏΜ·ΦΝ, άϊ”Οœ©ΧΰΗ¥Ζ÷ΫβΒΡΙΊΜΖΖ¥”ΠΆξ≥… staple ΒΡΙΙΫ®ΓΘ―–ΨΩ±μΟς, RCM Ζ¥”ΠΡή”κΙΧœύΚœ≥…ΕύκΡ≤Ώ¬‘œύΤΞ≈δ, ‘Ύ “Έ¬œ¬, ΐ–Γ ±±ψΩ…“‘ Βœ÷Άξ»ΪΉΣΜ·, ”≈”ΎΚήΕύ–η“Σ≥Λ ±ΦδΦ”»»«“ΉΣΜ·¬ ΒΆœ¬ΒΡΧΦ−ΧΦ≈ΦΝΣΖ¥”Π[13, 14]ΓΘStaple ΒΡΙΙΫ® Ήœ»–η“Σ‘ΎΕύκΡ–ρΝ–÷–“ΐ»κΝΫΗωΜρ’ΏΝΫΗω“‘…œΨΏ”– ΠΝ-ΥΪ»Γ ¥ζ ΒΡΖ« Χλ »ΜΑ± ΜυΥα ΉςΈΣΜΖΚœΥυ –η ΒΡΙΊ ΦϋΤω Ωι(building block)ΓΘ’β–©Ζ«Χλ»ΜΑ±ΜυΥαΒΡΧΊΒψ‘Ύ”Ύ ΠΝ-ΈΜΒΡ»Γ¥ζΜυ÷–“ΜΑψ“ΜΗωΈΣΦΉΜυ, Νμ“ΜΗωΈΣΝ¥œ©Μυ (Έλœ©ΜυΓΔ–Νœ©ΜυΒ»)ΓΘΨ≠ΙΐΙΧœύκΡΚœ≥… ÷ΕΈΫΪΥυ–ηΑ±ΜυΥαΫχ––÷π“ΜΥθΚœΒΟΒΫΡΩ±ξΕύκΡΝ¥Κσ, ΧΊΕ®ΈΜ÷Ο…œœύ ”ΠΒΡΝΫΗωΖ«Χλ»ΜΑ±ΜυΥα≤–ΜυΒΡ≤ύΝ¥ (Ν¥œ©Μυ) Ά®Ιΐ RCM Ζ¥”ΠΖΔ…ζΖ÷Ή”ΡΎΒΡœ©ΧΰΗ¥Ζ÷ΫβΖ¥”ΠΆ―»Ξ“ΜΖ÷Ή”““œ©–Έ≥…¥σΜΖœ©«≈, ”…”ΎΗΟΜΖœ©«≈œώΕ© ιΕΛ“Μ―υΥχΉΓ‘≠±ΨΙΙœσ“Ή±δΒΡ¬ί–ΐκΡ, Υυ“‘–ΈœσΒΡ≥Τ÷°ΈΣΕ© ικΡ (stapled peptide), Εχœύ”ΠΒΡΗΟΜΖœ©«≈Ω…≥Τ÷°ΈΣΕ© ι«≈ (staple)ΓΘ”…”ΎΖ«Χλ»ΜΑ±ΜυΥαΒΡ ΠΝ-ΥΪ»Γ¥ζ“‘ΦΑΕ© ι«≈ΒΡ–Έ≥…, ¥σΖυΕ»ΧαΗΏΝΥ ΠΝ-¬ί–ΐΙΙœσΕύκΡΒΡΈ»ΙΧ–‘[15]ΓΘ≤Δ«““Μ–©ΝΫ«Ή¬ί–ΐΕύκΡΖ÷Ή”ΒΡ¥©ΡΛΡήΝΠ“≤ΒΟΒΫΝΥΧα…ΐ, Ά§ ±Κή¥σ≥ΧΕ»…œΩΥΖΰΝΥ φ’≈…λ’ΙΉ¥Χ§œ¬ΒΡΕύκΡΝ¥“Ή ήΒΫκΡΟΗΥ°ΫβΤΤΜΒΒΡ»θΒψ[16]ΓΘ
1.1.2 ΙΊΦϋΓΑΤωΩιΓ±ΒΡΙΙΫ®
Κœ≥…Ε© ικΡΒΡΙΊΦϋΤωΩι“ΜΑψ «ΨΏ”–ΠΝ-œ©Χΰ≤ύΝ¥ΒΡΖ«Χλ»ΜΑ±ΜυΥα (ΆΦ2A), ΡΩ«ΑΒΡ―–ΨΩ÷ς“Σ≤…”Ο ΠΝ-C ΒΡΨχΕ‘ΙΙ–ΆΈΣ S Μρ R ΒΡ ÷–‘œ©ΜυΖ«Χλ»ΜΑ±ΜυΥα, Φρ≥ΤΈΣ Sn Μρ Rn (n ΈΣ≤ύΝ¥œ©Μυ÷–ΧΦΒΡΗω ΐ)ΓΘ¥ΥΆβ, ΜΙ”–”Ο”ΎΖλΚœκΡΗΡ‘λΒΡ ΠΝ-ΥΪœ©Μυ»Γ¥ζΑ±ΜυΥα, »γ B5[4]ΓΘΆ®ΙΐΗΏ–ßΓΔΒΆΚΡΓΔΦρ±ψΒΡ”–ΜζΚœ≥…ΖΫΖ®÷Τ±Η…œ ωΒΡΙΊΦϋΤωΩι «Ε© ικΡΚœ≥…ΒΡ±Ί“ΣΧθΦΰΓΘΙζΡΎΆβ–μΕύΩΈΧβΉι“‘≤ΜΆ§≤Ώ¬‘Ϋ®ΝΔΝΥ≤ΜΕ‘≥ΤΚœ≥… ÷–‘Α±ΜυΥαΖΫΖ®, ΡΩ«Α–ßΙϊΫœΚΟΒΡΖΫΖ®Αϋά®: “‘΅fύΚΆΣ (oxazinone) ΈΣ ÷–‘≈δΧε, Ά®Ιΐ≤ΜΕ‘≥ΤΆιΜυΜ·Ζ¥”ΠΚœ≥…ΡΩ±ξΜ·ΚœΈο[17−19]; Ά®Ιΐ ÷–‘≈δΧε BPB (benzylprolylaminobenzophenone) ”κΫπ τΡχΓΔ±ϊΑ±Υα (Η Α±Υα) Ζ¥”Π…ζ≥…Η¥ΚœΒΉΈο, Ψ≠Ιΐ ÷–‘”’ΒΦΚœ≥…ΗΏΕ‘”≥―Γ‘ώ–‘ΒΡ ÷–‘Α±ΜυΥα[20−22]ΓΘ”…”Ύ B5 «Ζ« ÷–‘Α±ΜυΥα,Κœ≥…œύΕ‘ΫœΈΣΦρ±ψ, Ω…≤…”Ο N-Εΰ±Ϋ―«ΦΉΜυΗ Α±ΥαθΞΈΣΤπ ΦΈο, ‘ΌΫχ––Η Α±ΥαθΞ ΠΝ-ΈΜΝΫ¥ΈΆιΜυΜ·Ζ¥”Π, ΉνΚσΨ≠ΙΐΥαΫβΓΔ»Ξ±ΘΜΛ±ψΩ…ΒΟΒΫœύ”ΠΒΡ ΠΝ-ΥΪ»Γ¥ζΑ±ΜυΥα(ΆΦ 2B)ΓΘΆ®Ιΐ…œ ωΖΫΖ®Κœ≥…ΒΟΒΫΒΡ”ΈάκΉ¥Χ§ SnΓΔRn ΚΆB5 ”κ Fmoc-OSu Μρ Fmoc-Cl Ζ¥”Π…ζ≥…œύ”ΠΒΡ Fmoc ±ΘΜΛΒΡΑ±ΜυΥαΚσΦ¥Ω…≤Έ”κ≥ΘΙφΒΡ Fmoc ≤Ώ¬‘ΕύκΡΚœ≥…ΓΘ
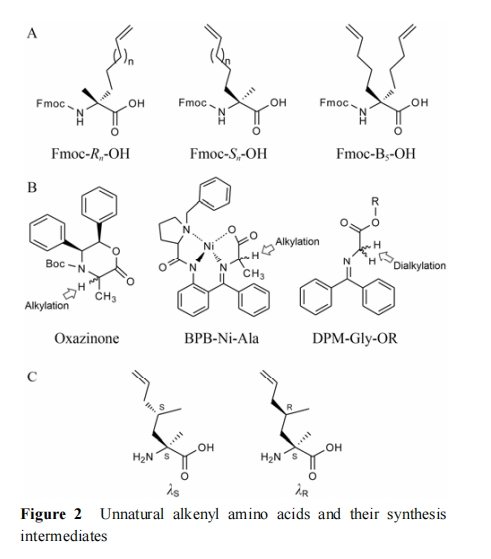
1.1.3 ΙΊΦϋΓΑΤωΩιΓ±ΒΡ«Ε»κΈΜΒψΒΡ―Γ‘ώΖΫΖ®
”…”Ύ≤ε»κΒΡΖ«Χλ»ΜΑ±ΜυΥα ΙΗΡ‘λΚσΒΡΕ© ικΡ–‘÷ ”κΧλ»ΜκΡ”–Υυ≤ν“λ, ‘Ύ≤Έ”κ PPIs ±Υυ±μœ÷≥ωΒΡΜν–‘“≤”–≤ΜΆ§ΓΘΈΣΝΥ…Η―Γ≥ωάμœκΒΡΗΏΜν–‘Ε© ικΡ, SnΓΔRn ΚΆ B5 ≤–Μυ«Ε»κΒΫΕύκΡ–ρΝ–ΒΡΈΜΒψ”»ΈΣΙΊΦϋΓΘΗυΨί“―±®ΒάΒΡ≤Ώ¬‘, ”–ΝΫ÷÷ΖΫΖ®ΫœΈΣ≥… λΓΘΒΎ“Μ÷÷≤Ώ¬‘ «Ά®ΙΐΖ÷ΈωΡΩ±ξΒΑΑΉΒΡ»ΐΈ§ΫαΙΙ ΐΨί, Ά®ΙΐΦΤΥψΜζΗ®÷ζΡΘΡβ”≈Μ·ΠΝ-¬ί–ΐκΡΤ§ΕΈ‘Ύ PPIs ÷–ΒΡΙΙœσ, ―Γ‘ώ≤Μ≤Έ”κ PPIs ΒΡΑ±ΜυΥα≤–ΜυΈΜΉςΈΣΖ«Χλ»ΜΑ±ΜυΥαΒΡ«Ε»κΈΜΒψ, Εχ“―÷ΣΒΡΜρ’Ώ «±ΜΆΤ≤βΩ…Ρή”κΑ–±ξœύΫαΚœΒΡΑ±ΜυΥα≤–Μυ”Π±Θ≥÷Άξ’ϊ, ΙΙΙΫ®ΚσΒΡΕ© ι«≈‘ΕάκΫαΚœΟφ, “‘±ήΟβ‘ΎΉς”Ο±μΟφ–Έ≥…Η…»≈[23]ΓΘΝμ“Μ÷÷≤Ώ¬‘ «’κΕ‘ΡΩ±ξκΡ, Ε‘ΤδΥυ”–ΡήΙΜΗΡΕ·ΒΡ–ρΝ–ΈΜΒψΫχ––Ζ«Χλ»ΜΑ±ΜυΥαΒΡΧφΜΜ, Κœ≥…“ΜœΒΝ–ΨΏ”–≤ΜΆ§Ε© ι«≈≤ε»κΈΜΒψΒΡΕ© ικΡ, ΉνΚσΆ®ΙΐΜν–‘…Η―ΓΒΡΖΫΖ®»ΖΕ®ΉνΦ―ΒΡ«Ε»κΈΜΒψ[24]ΓΘ÷ΒΒΟ÷Η≥ωΒΡ «Υδ»Μ‘Ύ“ΜΑψ«ιΩωœ¬”Π±ήΟβ‘ΎΕύκΡΚΆ ήΧεΒΡΉς”ΟΟφ¥ΠΙΙΫ®Ε© ι«≈, ΒΪ‘Ύ“Μ–©“‘ ΠΝ-¬ί–ΐ÷–ΒΡ ηΥ°Α±ΜυΥα (»γ ValΓΔLeuΓΔIle) ΫιΒΦ”κ ήΧεœύΜΞΉς”ΟΒΡ«ιΩωœ¬, ‘ΎΉς”ΟΟφ≤ύΙΙΫ®»ΪΧΦ«βΝ¥Ε© ι«≈Ω…“‘¥ζΧφ ηΥ°Α±ΜυΥα«Ε»κ ήΧεΒΡ ηΥ°≤έ÷–¥”Εχ ΙΕ© ι«≈ΖΔΜ”Έ»Ε®¬ί–ΐΚΆ≤Έ”κΫαΚœΒΡΥΪ÷ΊΙΠΡή[25−30]ΓΘSpeltz Β»[31]ΗϋΫχ“Μ≤ΫΆ®Ιΐœρ«≈ΆΖΑ±ΜυΥαΒΡœ©Μυ≤ύΝ¥÷–“ΐ»κΦΉΜυ»Γ¥ζΜυΡΘΡβ ηΥ°Α±ΜυΥα (ΆΦ 2C), Ά®ΙΐΕ‘ΦΉΜυ»Γ¥ζΜυΙΙ–ΆΒΡ”≈―Γ ΙΒΟΤδ”κ ήΧεΒΡΫαΚœΡήΝΠΫχ“Μ≤ΫΧαΗΏΓΘ
ΗυΨί ΠΝ-¬ί–ΐΫαΙΙΒΡΧΊΒψ, Άξ’ϊΒΡ¬ί–ΐ“Μ÷ή”– 3.6ΗωΑ±ΜυΥα≤–Μυ, ‘Ύ¥ΥΫαΙΙ÷–, i, i+4, i+7 ΚΆ i+11 ΈΜΒψΒΡ≤ύΝ¥¥Π”Ύ¬ί–ΐΒΡΆ§≤ύ (ΆΦ 3A)ΓΘΕ© ικΡΗΡ‘λ≤Ώ¬‘άϊ”Ο’β“ΜΧΊΒψ, Ά®ΙΐΈΜ”ΎΆ§≤ύΒΡ ΠΝ-C ≤ύΝ¥Ϋχ–– RCM Ζ¥”Π–Έ≥…ΜΖœ©«≈ΓΘΡΩ«Α, Ψ≠ΒδΒΡΗΡ‘λΖΫΖ® «‘Ύ i, i+4 ΈΜΒψΖ÷±π“‘ΝΫΗω S ΙΙ–ΆΈλœ©Μυ±ϊΑ±Υα (S5) ΧφΜΜ‘≠”–Α±ΜυΥα≤–Μυ≤Δ–Έ≥… staple, ¥”Εχ Βœ÷ΙΧΕ® ΠΝ-¬ί–ΐ“Μ÷ή[11](ΆΦ 3B); »γ”ΟœύΥΤΒΡΖΫΖ®‘Ύ i, i+7 ¥Π–Έ≥… staple ‘ρΩ…“‘ΙΧΕ®¬ί–ΐΝΫ÷ή, œύ”ΠΝΫΈΜΒψ¥Π‘ρ–η“Σ≤ύΝ¥Ηϋ≥ΛΒΡR8 ΚΆ S5 Μρ S8 ΚΆ R5 ΒΡΉιΚœ (ΆΦ 3C)ΓΘ≥ΐΝΥ…œ ωΝΫ÷÷Ψ≠ΒδΖΫΖ®Άβ, i, i+3 ΈΜΒψ“≤Ω… ”ΟΕ© ικΡΗΡ‘λ, ΒΪ–η“ΣΉΔ“βΒΡ « staple ΒΡ≥ΛΕ»ΨΏ”–“ΜΕ®ΒΡΝιΜν–‘: Β±ΤδΈΣ 8 ΗωΧΦ‘≠Ή” ±, –η“Σ R5 ”κ S5 ΒΡΫαΚœ (ΆΦ 3D); Β±ΤδΈΣ 6 ΗωΧΦ‘≠Ή” ±, ‘ρ–η“Σ R5”κ S3Μρ’Ώ R3”κ S5ΒΡΉιΚœ≤≈Ω…“‘Έ§≥÷Κœ ΒΡΕύκΡΙΙœσ[32, 33]ΓΘ
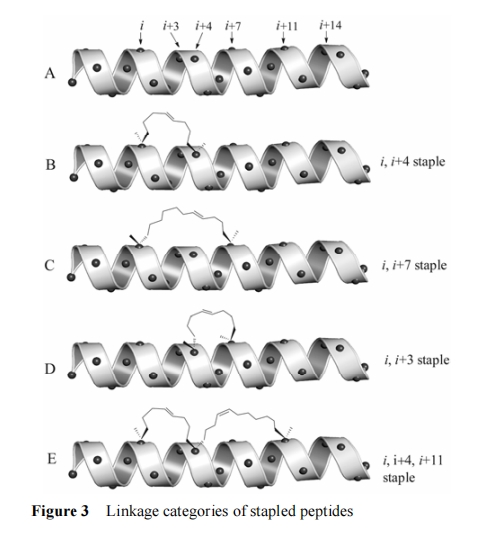
2014 Ρξ Verdine ΩΈΧβΉι”÷±®ΒάΝΥΝν»ΥΕζΡΩ“Μ–¬ΒΡΓΑΖλΚœκΡΓ±(stitched peptides), «…ΟνΒΊ…ΐΦΕΝΥ‘≠”–ΒΡΕ© ικΡΗΡ‘λΖΫΖ®ΓΘΆ®ΙΐΫΪ ΠΝ ΈΜΥΪΈλœ©Μυ»Γ¥ζΒΡΖ« ÷–‘Α±ΜυΥα B5 “ΐ»κΕύκΡ–ρΝ–, ΤδΝΫ≤ύ‘ΌΗς“ΐ»κ“ΜΗω≈ΦΝΣΖ¥”Πœύ ”ΠΒΡ Sn ΚΆ Rn (»γ S5+B5+R5ΓΔS5+B5+S8ΓΔR8+B5+S8Β»), ¥”Εχ‘Ύ i, i+4, i+8Μρ i, i+4, i+11 Μρ i, i+7, i+14 ¥Π“‘“ΜΗωΙ≤”Ο÷–Φδ«≈ΆΖΒΡΖΫ Ϋ–Έ≥…ΥΪ÷ΊΕ© ι«≈, ”׻㥩’κ“ΐœΏ“ΜΑψΫΪ¬ί–ΐκΡΝ¥ΖλΚœΤπά¥, ¥”ΕχΩ…“‘¥σΖυΕ»ΒΊΧαΗΏΝΥΕ© ικΡ ΠΝ-¬ί–ΐ–‘ (ΆΦ 3E)[4]ΓΘ
ΡΩ«Α‘Ύ i, i+4 ΈΜΒψ“‘ S5+S5 ΈΣ«Ε»κΤωΩιΒΡΕ© ικΡΗΡ‘λΖΫΖ®‘ΎΙζΡΎΆβΩΈΧβΉι÷–”Π”ΟΉνΈΣΙψΖΚΓΘΕ‘”ΎΕΧ κΡά¥Ϋ≤, Ω…“‘Ά®Ιΐ“ΜΗωΕ© ι«≈Άξ≥… ΠΝ-¬ί–ΐΒΡΙΧΕ®, »γΙϊœ»ΒΦΈοΈΣ≥ΛκΡ, ‘ρΩ…“‘Ά®ΙΐΝΫΗωΜρ’ΏΝΫΗω“‘…œΒΡΕάΝΔ staple Ϋχ––ΫαΙΙΙΧΕ® (ΥΪΕ© ικΡ, ≤ΜΆ§”Ύ stitched peptides)[34]ΓΘΒΪ «–η“ΣΉΔ“βΒΡ «, “‘ R5+R5 ¥ζΧφ S5+S5‘Ύ i, i+4 ΈΜΒψ…œΫχ––ΗΡ‘λΒΡ―–ΨΩ‘ρΖ«≥Θ…ΌΦϊ, ÷ς“Σ“ρΈΣΆ®Ιΐ R5+R5 ΒΡΗΡ‘λ≤ΜΒΪ≤ΜΡήΙΜΧαΗΏΡΩ±ξκΡΒΡ ΠΝ-¬ί–ΐ–‘, Ζ¥ΕχΜα ΙΤδ”–Οςœ‘ΒΡœ¬ΫΒ, œΗΑϊ…ψ»ΓΡΩ±ξκΡΒΡΡήΝΠ“≤Υφ÷°Φθ»θ[35]ΓΘ
1.1.4 »ΪΧΦΝ¥Ε© ικΡΒΡΙΧœύΚœ≥…
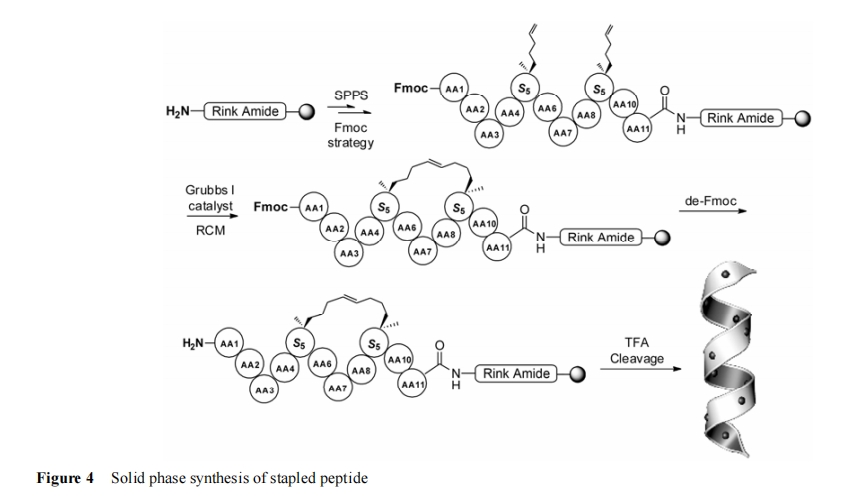
Ε© ικΡΒΡΚœ≥…”κ“ΜΑψΒΡΕύκΡΙΧœύΚœ≥…Μυ±Ψ“Μ÷¬, ΫœΈΣΧΊ±πΒΡ « Sn ΜρRn ‘Ύ“ΐ»κ ±, ”…”ΎΈΜΉηΫœ¥σ–η“Σ»γ PyAOP Β»ΫœΈΣΗΏ–ßΒΡΥθΚœΦΝ»Ζ±ΘΥθΚœΖ¥”ΠΒΡΝ¥Ϋ”–߬ ΓΘ”…”ΎΗΟ ΠΝ-ΥΪ»Γ¥ζΑ±ΜυΥαΒΡ”ΈάκΑ±ΜυΈόΖ®Ά®Ιΐ≥ΘΙφΒΡ Kaiser ήα»ΐΆΣΖΫΖ®Ϋχ––Φύ≤β, Ω…‘Ύœύ”ΠΈΜ÷ΟΒΡΑ±ΜυΥαΥθΚœΆξ≥…Κσ»Γ…ΌΝΩΫχ––Ν―Ϋβ≤Δ≤…”Ο LC/MS ΖΫΖ®Ϋχ––»Ζ÷ΛΓΘΈΣ»Ζ±ΘRCM Ζ¥”Π‘ΎΒΞΧθΕύκΡΖ÷Ή”ΡΎΖΔ…ζ, ΜΖΚœΖ¥”Π–η“Σάϊ”ΟΙΧœύΚœ≥…ΒΡΦΌœΓ Ά–ß”Π[36]‘ΎΙΧœύ ς÷§…œΫχ––ΓΘ’ϊΗωΕ© ικΡΒΡΚœ≥…÷Τ±ΗΙΐ≥Χ¥σ÷¬Ζ÷ΈΣκΡΝ¥ΒΡΚœ≥…ΓΔ≤ύΝ¥ΒΡ RCM Ζ¥”ΠΓΔN-ΕΥΒΡ–ό ΈΓΔ‘ΊΧεΒΡ«–≥ΐΓΔΕ© ικΡΒΡ¥ΩΜ·Β»ΦΗΗω≤Ϋ÷η (ΆΦ 4), “―Ψ≠≥…ΈΣΫœΈΣ≥… λΒΡΖΫΖ®[37]ΓΘ
1.2 »ΪΧΦ«βΝ¥Ε© ικΡΒΡΜν–‘ΗΡ‘λΈο―–ΨΩΫχ’Ι
1.2.1 P53-MDM2/MDMX œύΜΞΉς”Ο“÷÷ΤΦΝ
p53Μυ“ρ «“Μ÷÷÷Ί“ΣΒΡ“÷Α©Μυ“ρ,”κDNAΒΡ–όΗ¥ΓΔœΗΑϊΖ÷Μ·ΓΔœΗΑϊ÷ήΤΎΒΡΒςΩΊΒ»…ζΟϋΙΐ≥ΧœΔœΔœύΙΊΓΘp53ΒΡ»± ßΚΆΆΜ±δΦΑœύΙΊΒΑΑΉΟΗΧεΒΡΫΒΫβΜαΒΦ÷¬÷ΉΝωœΗΑϊΒΡ≤ζ…ζΓΘMDM2ΦΑMDMX «p53ΒΡ÷ς“Σ“÷÷Τ“ρΉ”,ΝΫ’ΏœύΜΞ–≠Ά§≤ΔΆ®Ιΐ≤ΜΆ§ΒΡ–≈Κ≈ΆΨΨΕ“÷÷Τp53ΒΡΜν–‘ΓΘE3ΖΚΥΊΝ§Ϋ”ΟΗMDM2Ω…”κp53ΒΑΑΉΒΡΖ¥ΦΛΜν”ρΫαΚœ,ΫιΒΦp53ΒΡΖΚΥΊΜ·¥”ΕχΫΒΒΆp53ΒΡΈ»Ε®–‘ΓΘMDMX‘ρ÷ς“ΣΆ®Ιΐ”κp53ΒΡΉΣ¬ΦΜν–‘«χΫαΚœ,“÷÷Τp53Ε‘Τδœ¬”ΈΜυ“ρΒΡΉΣ¬ΦΜν–‘,¥”Εχ≤ζ…ζ“÷÷ΤΉς”Ο[38,39]ΓΘ―–ΨΩΖΔœ÷,p53-MDM2ΒΑΑΉΦδΒΡœύΜΞΉς”Ο ±,“ΜΕΈΈΜ”Ύp53NΕΥΒΑΑΉΖ¥ΦΛΜν”ρΒΡ≥Λ15ΗωΑ±ΜυΥα≤–ΜυΒΡΠΝ-¬ί–ΐ(LSQETFSDLWKLLPEN)”κMDM2ΒΡ ηΥ°ΑΦ≤έΫαΚœ≤ΔΒςΩΊΤδœύΜΞΉς”Ο[40],Τδ÷–Phe19ΓΔTrp23ΚΆLeu26 «”κMDM2œύΫαΚœΥυ±Ί–κΒΡ[30,41]ΓΘVerdineΩΈΧβΉι[42]άϊ”Ο»ΪΧΦ«β≤ύΝ¥ΒΡstapleΗΡ‘λ≤Ώ¬‘,Ά®ΙΐΕ‘Μ·ΚœΈοœ©ΜυΑ±ΜυΥα“ΐ»κΈΜ÷ΟΒΡ…Η―Γ,ΫαΚœΕ‘Υα–‘ΦΑΦν–‘Α±ΜυΥα≤–ΜυΒΡΧφΜΜΗΡ±δΕύκΡΨΜΒγΚ…“‘‘ωΦ”¥©ΡΛ–‘Β»≤Ώ¬‘,Κœ≥…≤Δ…Η―Γ≥ωΝΥΨΏ”–ΗΏ«ΉΚΆΝΠΓΔΗΏΠΝ-¬ί–ΐ–‘“‘ΦΑ¥©ΡΛΡήΝΠΒΡΕ© ικΡSAH-p53-8(Ac-QSQQTF[R8NLWRLLS5]-QN-NH2(±ΨΈΡΈΣΉΦ»Ζ±μ ωΕ© ικΡΒΡΗΡ‘λΫαΙΙΈΜ÷Ο,”ΟΚΎΧε±ξ≥ωΧφΜΜΒΡΖ«Χλ»Μœ©ΜυΑ±ΜυΥα≤Δ”ΟΓΑ[Γ≠]Γ±±μ ΨΈΜ”ΎΕ© ι«≈¥σΜΖΡΎΒΡΑ±ΜυΥα–ρΝ–),ΤδΠΝ-¬ί–ΐ–‘¥οΒΫ85%ΓΔKD÷Β¥οΒΫ55nmolΓΛL−1,≤ΔΩ…“‘ΆΗΙΐœΗΑϊΡΛ“ΐΤπ÷ΉΝωœΗΑϊΒρΆωΓΘΥφΚσVerdineΩΈΧβΉι[43]…ν»κ―–ΨΩΖΔœ÷SAH-p53-8ΜΙΩ…“‘”––ßΒΊ“÷÷ΤMDMX,Ε‘”ΎMDMXΒΡ«ΉΚΆΝΠ±»MDM2ΗΏ25±ΕΓΘ“ρ¥ΥSAH-p53-8”–œΘΆϊΉςΈΣ“÷÷Τ÷ΉΝωΒΡΥΪΑ–œρ“÷÷ΤΦΝΫχ––≥…“©–‘―–ΨΩΓΘ‘ΎΥφΚσΒΡΫχ“Μ≤Ϋ”≈Μ·―–ΨΩ÷–[44],Ά®Ιΐ …ΨζΧε’Ι ΨΦΦ θΕ‘ΗΟSAH-p53-8œ»ΒΦΈο–ρΝ–Α±ΜυΥαΈΜΒψΆΜ±δΒΟΒΫΝΥ“ΜΧθ–¬ΒΡΨΏ”–ΗϋΗΏMDMX/MDM2«ΉΚΆΝΠΒΡΕύκΡAc-LTFEHYWAQLTS-NH2,Ψ≠ΙΐΕ‘Glu4ΚΆThr11Ζ÷±π≤…”Ο R8 ΚΆ S5 ΧφΜΜ≤Δ–Έ≥…Ε© ικΡ“‘ΦΑΕ‘ Asn5ΓΔLeu10 ΒΡ≤–ΜυΈΜΒψΧφΜΜΚΆ C ΕΥΫαΙΙ”≈Μ·ΒΟΒΫΝΥΕ© ικΡΚρ―ΓΈο ATSP-7041 (Ac-LTF[R8EYWAQCbaS5]SAANH2), Ε‘ MDM2 ΚΆ MDMX ”κ p53 ΒΡΫαΚœ“÷÷Τ≥Θ ΐ(Ki) Ζ÷±π¥οΒΫ 0.9 nmolΓΛL−1 ΚΆ 6.8 nmolΓΛL−1 Υ°ΤΫ, ‘ΎΧεΡΎΆβΨυœ‘ ΨΝΥœ‘÷χΒΡ÷ΉΝω“÷÷ΤΜν–‘«““©¥ζ–‘÷ ΝΦΚΟ, Ά®ΙΐΕ‘Κρ―ΓΈοΫχ“Μ≤Ϋ”≈Μ·ΒΟΒΫΒΡΕ© ικΡ ALRN- 6924 ΡΩ«Α“―Ϋχ»κ II ΤΎΝΌ¥≤―–ΨΩΫΉΕΈ (FDA ΝΌ¥≤―–ΨΩ±ύΚ≈ NCT02264613), ”–Άϊ…œ –≥…ΈΣ ΉΗωΕ© ικΡ÷ΈΝΤ“©ΈοΓΘ
1.2.2 BCL-2 ΒΑΑΉΦ“ΉεΒΡΒςΩΊ
œΗΑϊΒρΆωœύΙΊ BCL-2ΒΑΑΉΦ“ΉεΖ÷ΈΣΝΫ¥σάύ, “ΜάύΨΏ”–“÷÷ΤœΗΑϊΒςΆωΉς”Ο, »γ BCL-2ΓΔBCL-XLΓΔBCL-1ΓΔMCL-1 Β»; Νμ“ΜάύΩ…¥ΌΫχœΗΑϊΒρΆω, »γ BAXΓΔBCL-XSΓΔBAKΓΔBID Β»[45]ΓΘ¥σΝΩ―–ΨΩΖΔœ÷ BCL-2 ΒΡΆ§‘¥ΫαΙΙ”ρ BH3 ÷–ΒΡΝΫ«Ή ΠΝ-¬ί–ΐΤ§ΕΈΡήΫαΚœΒΫ”… BH1ΓΔBH2 ΚΆ BH3 Ι≤Ά§–Έ≥…ΒΡΩΙΒρΆωΫαΙΙ”ρΒΡ ηΥ°«χ÷–, ¥”ΕχΖΔΜ”¥ΌΫχœΗΑϊΒρΆωΒΡΉς”Ο[46]ΓΘWalensky Β»[12]Ά®ΙΐΡΘΡβ BID ΒΡ BH3 «χ”ρ ΠΝ-¬ί–ΐΤ§ΕΈ (EDIIRNIARHLAQVGDSNLDRSIW), …ηΦΤΚœ≥…ΝΥ“ΜΉιΨΏ”–Έ»Ε® ΠΝ-¬ί–ΐΫαΙΙΒΡΕ© ικΡ SAHBs, Τδ÷–…Η―Γ≥ωΝΥ‘Ύ¥©ΡΛΡήΝΠΓΔΫαΚœΡήΝΠΚΆΦΛΜνœΗΑϊΒρ ΆωΜν–‘÷–ΨΏ”–ΩΣΖΔ«±ΝΠΒΡΕ© ικΡ SAHBA (EDIIRNIARHLA[S5VGDS5]NLDRSIW)ΓΘΤδ KD ÷Β”…‘≠–ΈΒΡ 269 nmolΓΛL−1 ΧαΗΏ÷Ν 38.8 nmolΓΛL−1, ΠΝ-¬ί–ΐ–‘ΈΣ 87.5%; SAHBA ‘ΎΧεΆβ―Σ«ε÷–ΒΡΑκΥΞΤΎ”…‘≠–ΈΒΡ 3.1 h ΧαΗΏ÷Ν 29.4 hΓΘΥφΚσ Stewart Β»[47]”÷…Η―ΓΝΥ BCL-2 ΫαΙΙ”ρ÷–ΒΡ“ΜœΒΝ–ΨΏ”– ΠΝ-¬ί–ΐΫαΙΙΒΡΤ§ΕΈ≤ΔΗΡΙΙ≥…ΈΣΕ© ικΡ, ΖΔœ÷ MCL-1 ÷–ΒΡ BH3 Τ§ΕΈ¬ί–ΐ±Ψ…μ (KALETLRRVGDGVQRNHETAF) ΈΣΒΞ“ΜΒΡ MCL-1 “÷÷ΤΦΝΓΘΨ≠ΙΐΗΡΙΙΒΡΕ© ικΡ MCL-1 SAHBX ÷–, MCL-1 SAHBD(KALETLRRVGDGV[S5RNHS5]TAF) ΈΣάμœκΒΡ“÷÷ΤΦΝ, Τδ KD÷ΒΈΣ 10Γά3 nmolΓΛL−1 (‘≠–ΈΈΣ 245 Γά29 nmolΓΛL−1)ΓΘΡΩ«Α“‘ΗΟΒΑΑΉΈΣΑ–ΒψΒΡΕ© ικΡάύœ»ΒΦΜ·ΚœΈο¥Π‘ΎΝΌ¥≤«Α―–ΨΩΫΉΕΈΓΘ
1.2.3 ΤδΥϊΧΦ«βΝ¥Ε© ικΡΒΡ―–ΨΩ«ιΩω
≥ΐΝΥ…œ ω―–ΨΩΫœ‘γ≤ΔΦ¥ΫΪ≥…“©ΒΡΕ© ικΡάύΜ·ΚœΈο“‘Άβ, ΥφΉ≈Ε© ικΡ≥…ΈΣΕύκΡ“©Έο―–ΨΩΒΡ»»Βψ, ―–ΨΩΖΔœ÷ΝΥΕύ÷÷ΒςΫΎΦ≤≤ΓœύΙΊ PPIs ΒΡΕ© ικΡ“©Έοœ»ΒΦΈοΓΘ±μ 1 ΉήΫαΝΥΡΩ«Α“―±®ΒάΒΡ”κΑ–ΒΑΑΉœύΜΞΉς”Ο»Ζ«–ΓΔΨΏ”–œ‘÷χ“©άμΜν–‘ΒΡΕ© ικΡΗΡΙΙΈοΒΡ―–ΨΩ«ιΩωΓΘ
2 ΤδΥϊάύΕ© ικΡΒΡΚœ≥……ηΦΤΦΑ”Π”Ο
‘Ύ…œ ω≤…”Ο RCM Ζ¥”ΠΈ»Ε® ΠΝ-¬ί–ΐΒΡΕ© ικΡ≤Ώ¬‘Έ ά÷°«Α, Ά®Ιΐ≤ύΝ¥ΦδΒΡ≈ΦΝ§Έ»Ε®ΕύκΡ ΠΝ-¬ί–ΐΙΙœσΒΡΙΛΉς“―”–ΚήΕύ±®Βά, άΐ»γ‘Ύ≤ύΝ¥Φδ–Έ≥…θΘΑΖΦϋ[101]ΓΔ≈ΦΒΣ±Ϋ[102]ΓΔκξ[103]ΓΔΕΰΝρΦϋ[104]Β», ΒΪ’β–©ΖΫΖ®ΗΡ‘λΚσΒΡΕύκΡ»¥≤Δ≤ΜΡή≥Τ÷°ΈΣΕ© ικΡ, ÷ς“Σ”…”ΎΥϋΟ«‘Ύ“©¥ζ―ß–‘÷ …œ≤Μ»γ»ΪΧΦ«βΝ¥Ε© ικΡΈ»Ε®[105]ΓΘΫϋΡξά¥“≤≥ωœ÷ΝΥ“Μ–©–¬ΒΡΗΡ‘λ≤Ώ¬‘, Υδ»ΜΥϋΟ«ΒΡΝ¥Ϋ”ΜΖ“≤”–‘”‘≠Ή”≤Έ”κ, ΒΪΤδ‘ΎΈ»Ε®ΙΙœσΓΔΧαΗΏΜν–‘ΚΆ“©¥ζΈ»Ε®–‘ΖΫΟφ”κΨ≠Βδ»ΪΧΦ«βΝ¥Ε© ικΡ”–“λ«ζΆ§ΙΛ÷°¥Π, Ω…“‘ΫΪΤδΙιΈΣΙψ“εΒΡΕ© ικΡΗΡ‘λΖΫΖ®ΓΘœ¬ΈΡΫΪΑ¥’’Ν§Ϋ”«≈ΜΖΒΡΫαΙΙΕ‘ΤδΫχ––Ζ÷άύΥΒΟςΓΘ
2.1 »ΐΏρ«≈ΜΖ
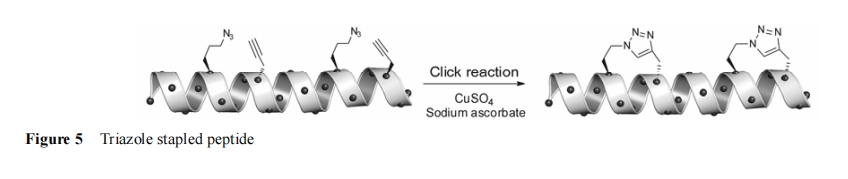
ΒψΜςΜ·―ß (click chemistry) ÷–ΒΡ Huisgen-1,3 ≈ΦΦΪΜΖΦ”≥…[106]Ζ¥”Π «‘ΎΆ≠¥ΏΜ·œ¬, »≤Μυ”κΒΰΒΣΜυΖ¥”Π…ζ≥…«χ”ρ―Γ‘ώ–‘ΒΡ 1,4-»Γ¥ζ»ΐΏρΓΘ”…”Ύ Click Ζ¥”ΠΨΏ”–ΗΏΖ¥”Π–‘ΓΔ―Γ‘ώ–‘ΚΆ…ζΈοœύ»ί–‘[107−111], ΤδΜΖΦ”≥…≤ζΈοΨΏ”–Ε‘ΒΑΑΉΥ°ΫβΟΗΒΡΈ»Ε®–‘[112]Β»”≈Βψ, CantelΒ»[113]±ψ¬ œ»≥Δ ‘ΫΪΗΟΜΖΦ”≥…Ζ¥”Π”Π”ΟΒΫΕύκΡ≤ύΝ¥ΒΡ≈ΦΝΣ≤Δ»ΓΒΟΝΥΝν»Υ¬ζ“βΒΡΫαΙϊΓΘΗΟΖΫΖ®‘ΎκΡΝ¥ΒΡ i, i+4 ΈΜΒψ¥ΠΖ÷±π“ΐ»κΝΥ ΠΝ-ΈΜΡ©ΕΥΨΏ”–»≤ΜυΚΆΒΰΒΣΜυΒΡΖ«Χλ»ΜΑ±ΜυΥα, ΉνΚσ¥ΩΜ·ΒΡκΡΝ¥‘Ύ“Κœύ÷–Ά®Ιΐ CuSO4”κΩΙΜΒ―ΣΥαΒΡ¥ΏΜ·ΖΔ…ζ clickΖ¥”Π–Έ≥…ΓΑ»ΐΏρ«≈Γ±(ΆΦ5)ΓΘάϊ”Ο click Ζ¥”ΠΗΡ‘λΒΡ PTHrP κΡ±»Τδ‘≠–ΈκΡΜν–‘ΧαΗΏΝΥ 5ΓΪ10 ±Ε, ≤Δ”κθΘΑΖ«≈ΗΡ‘λΚσΒΡ PTHrP κΡ”–Ή≈œύΥΤΒΡ…ζΈοΜν–‘[114]ΓΘΥφΚσ Kawamoto Β»[115]‘Ύ…ηΦΤΚœ≥… BCL-9 Ε© ικΡ ±“≤”Π”Ο click Ζ¥”Πά¥ΙΙΫ®»ΐΏρ«≈, Ά®Ιΐ«Ε»κΈΜΒψΒΡ…Η―Γ“‘ΦΑ»ΐΏρ«≈≥ΛΕ»ΒΡ±δΜΜ”≈Μ·≥ωάμœκΒΡΕ© ικΡΓΘΧΊ±π «‘ΎΗΟκΡΝ¥…œ“ΐ»κΒΡΥΪ»ΐΏρ«≈ΜΖ ΙΒΟΗΡ‘λΚσΒΡΕ© ικΡ ΠΝ-¬ί–ΐΚ§ΝΩ¥σ”Ύ 90%, ΉνΗΏ…θ÷Ν¥οΒΫ 99%, Τδ«ΉΚΆΝΠ“‘ΦΑ¥ζ–ΜΈ»Ε®–‘”κ“Α…ζ–Ά BCL9œύ±»ΕΦ”–Κή¥σΒΡΧαΗΏΓΘ
œύ±»»ΪΧΦ«βΝ¥Ε© ικΡ, »ΐΏρΕ© ικΡΒΡ–Έ≥…Υυ–η“ΣΒΡΆ≠¥ΏΜ·ΦΝΖ«≥ΘΝ°Φέ«“ΕΨ–‘ΫœΒΆ, «“Μ÷÷Ω…–––‘ΫœΗΏΒΡΕ© ικΡΙΙΫ®ΖΫΖ®, ΒΪΡΩ«ΑΕ‘”ΎΥϋ‘Ύ ΠΝ-¬ί–ΐ–‘“‘ΦΑΟΗΫβΈ»Ε®–‘…œΗΡ…ΤΉς”Ο»‘–η…ν»κ―–ΨΩΓΘ
2.2 Νρ¥ζ«≈ΜΖ
‘ΎΕύκΡΜρ’ΏΒΑΑΉ÷ ΒΡ–ό ΈΗΡ‘λ÷–,ΑκκΉΑ±ΥαΒΡ―ή…ζΜ·“―≥…ΈΣ“Μ÷÷±Ί≤ΜΩ……ΌΒΡΙΙΫ®ΖΫ Ϋ,“≤ «Μ·―ß…ζΈο―ß―–ΨΩΒΡ÷Ί“ΣΖΫΖ®ΓΘΥδ»ΜΡΩ«ΑΗΡ‘λΑκκΉΑ±ΥαΒΡΖΫΖ®”–ΆιΜυΜ·ΓΔΙ≤ινΦ”≥…ΓΔ―θΜ·ΚΆΜΙ‘≠Β»ΖΫΖ®[116,117],‘ΎκΡΝ¥…œΆ®Ιΐ≤ύΝ¥έœΜυ÷°Φδ≈ΦΝ§ΙΙΫ®«≈ΜΖά¥Έ»Ε® ΠΝ-¬ί–ΐΒΡΖΫΖ®¥σΕύΨ÷œό”Ύ–Έ≥…ΕΰΝρΦϋΓΘ»ΜΕχΒ±ΕΰΝρΦϋ¥Π”Ύ―θΜ·ΜρΜΙ‘≠ΧθΦΰ ±Τδ–‘÷ ≤ΜΙΜΈ»Ε®ΓΘάϊ”ΟΤδΜν–‘έœΜυ”κΤδΥϊ¥ζ–ΜœύΕ‘Έ»Ε®ΒΡΝ§Ϋ”ΫαΙΙΖ¥”ΠΒΟΒΫΒΡ«≈ΜΖ±»ΕΰΝρΦϋΒΡΈ»Ε®–‘«Ω, ”…¥Υ“≤―ή…ζ≥ωœ¬ ωΕύ÷÷–¬–ΆΕ© ικΡΙΙΫ®ΖΫΖ®ΓΘ
2.2.1 Νρ¥ζΦΉΜυΝΣ±Ϋ«≈ΜΖ
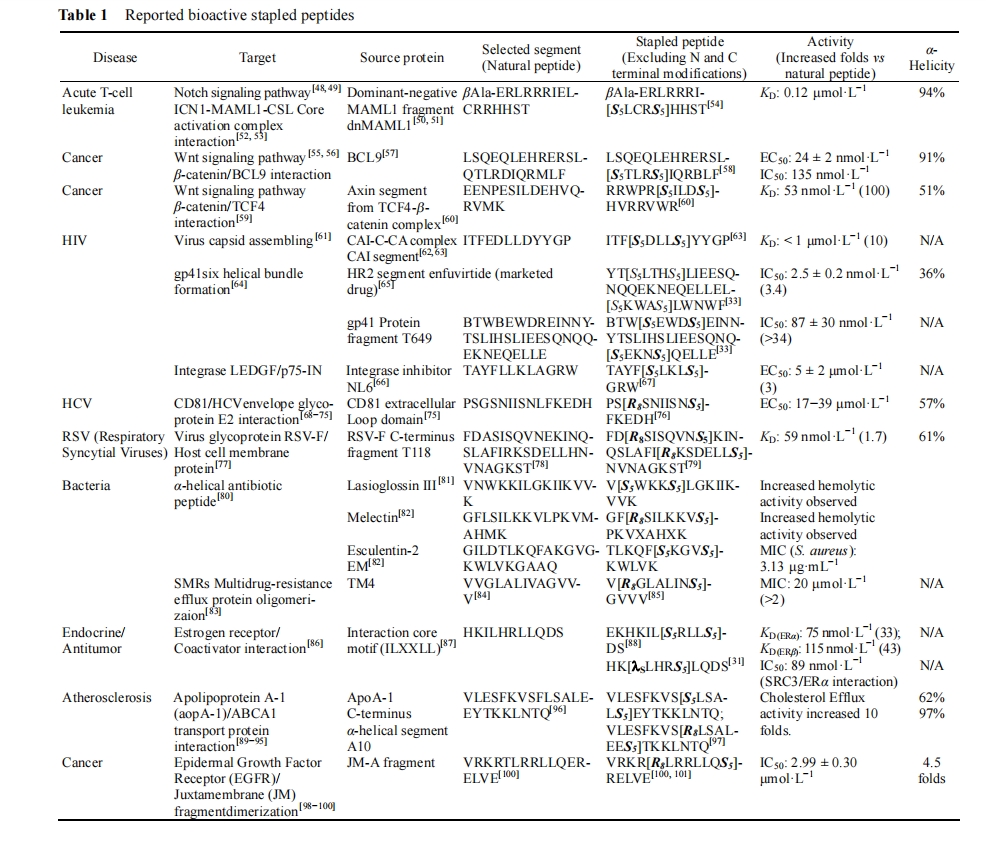
Muppidi Β»[118]±®ΒάΝΥάϊ”Ο 4,4'-ΕΰδεΦΉΜυΝΣ±Ϋ (Bph) Μρ 6,6'-ΕΰδεΦΉΜυ-2,2'-ΝΣΏΝύΛ (Bpy) ΉςΈΣΝ§Ϋ”±έ, Ά®Ιΐ”κ i, i+7 ΈΜΒΡΑκκΉΑ±Υα≤ύΝ¥Ζ¥”Π–Έ≥…Νρ¥ζΦΉΜυΝΣ±ΫΜρΝΣΏΝύΛ«≈ΜΖ (ΆΦ 6)ΓΘΥϊΟ«ΥφΚσΫΪΗΟΗΡ‘λ≤Ώ¬‘‘Υ”ΟΒΫΨΏ”– p53-MDM2/MDMX œύΜΞΉς”ΟΥΪ÷Ί“÷÷ΤΉς”ΟΒΡΕύκΡ–ρΝ– (LTFEHYWAQLTS) ΗΡ‘λ÷–ΓΘ”…”Ύ’βΕΈ–ρΝ–÷–≤ΜΚ§”–ΑκκΉΑ±Υα, “ρ¥ΥΫΪ±©¬Ε‘Ύ»ήΦΝ≤ύΒΡ Glu4 ΚΆ Thr11ΧφΜΜΈΣΑκκΉΑ±Υα‘ΌΫχ––«≈ΜΖΒΡΙΙΫ®ΓΘΆ®ΙΐΜν–‘…Η―Γœ‘ ΨΗΡΙΙΚσΒΡΕ© ικΡ≤ΜΒΪ ΠΝ-¬ί–ΐΚ§ΝΩΚΆ…ζΈοΜν–‘”–ΗΡ…ΤΕχ«“¥©ΡΛΡήΝΠΒΟΒΫΝΥ¥σΖυΕ»ΒΡΧαΗΏΓΘ
Muppidi Β»[119]”÷‘Υ”ΟΝρ¥ζΦΉΜυΝΣ±Ϋ≤Ώ¬‘Ε‘ StichtΒ»[61]±®ΒάΒΡ“÷÷Τ HIV-1 ≤ΓΕΨ“¬Ω«ΒΡΉιΉΑΒΡ CAI ΒΑΑΉΤ§ΕΈ (ITFEDLLDYYGP) Ϋχ––ΗΡΙΙ, ΫΪ»ήΦΝ≤ύΒΡ Glu4ΚΆ Gly11 ΧφΜΜΈΣΑκκΉΑ±ΥαΚσΫχ––«≈ΜΖΒΡΙΙΫ®ΓΘΕ‘ΗΡΙΙΈοΜν–‘≤β ‘œ‘ Ψ, Τ䥩ΡΛΡήΝΠΕΦ”–ΦΪ¥σΒΡΧαΗΏ, «“‘ΎœΗΑϊΥ°ΤΫΒΡ≤ΓΕΨΗ–»Ψ Β―ι÷–œ‘ Ψ≥ωΗΏ“÷÷ΤΜν–‘ΓΘ”» Τδ «…Η―Γ≥ω“ΜΧθΦ»ΡήΙΜ”κ HIV-1 “¬Ω«ΒΑΑΉΧΦΕΥΫαΚœ(KD =17.5 ΠΧmolΓΛL−1) ”÷Ρή”κ gp120 ΒΑΑΉΫαΚœ (KD =7.4 ΠΧmolΓΛL−1) ΒΡΗΡΙΙΕ© ικΡ (ISF[CELLDYYC]ESGS), ΥΒΟςΤδΨΏ”–“÷÷Τ“¬Ω«ΉιΉΑΚΆ“÷÷Τ HIV-1 ≤ΓΕΨΒΡΫχ»κœΗΑϊΒΡΥΪ÷Ί“÷÷ΤΉς”ΟΓΘ
ά¥Ή‘ BH3 ΒΑΑΉ÷–ΒΡΕύκΡ NoxaB-(75−93)-C75A(AAQLLRIGDKVNLRQKLLN) ΡήΙΜΗΏ―Γ‘ώ–‘ΒΡ”κMcl-1 ΒΑΑΉΫαΚœ¥”Εχ≤ζ…ζ“÷÷Τ MCL-1 ΒΡΉς”Ο[120]ΓΘMuppidi Β»[121]ΈΣΝΥ”≈Μ·ΕύκΡ Noxa BH3 ΒΡΜν–‘, Ά®ΙΐΝρ¥ζΦΉΜυΝΣ±Ϋ«≈ΜΖ≤Ώ¬‘, ΫΪ Gln77”κ Lys84ΧφΜΜΈΣΑκκΉΑ±Υα (AACLLRIGDCVNLRQKLLN) ‘ΌΫχ–– staple ΙΙΫ®ΓΘΥφΚσΕ‘œΒΝ–ΗΡΙΙΈοΫχ––…ζΈοΜν–‘≤β ‘, ΫαΙϊœ‘ ΨΨ≠ΙΐΗΡΙΙΚσΒΡΕ© ικΡΒΡ ΠΝ-¬ί–ΐΚ§ΝΩΓΔ¥©ΡΛΡήΝΠ“‘ΦΑΟΗΫβΈ»Ε®–‘ΕΦ”–¥σΖυΕ»ΧαΗΏ, Εχ«“Ε‘ MCL-1 ΙΐΕ»±μ¥οΒΡ U937 œΗΑϊ”–ΚήΚΟΒΡ“÷÷ΤΜν–‘ΓΘ
2.2.2 Νρ¥ζΖζ±Ϋ«≈ΜΖ
SpokoynyΒ»[122]“‘»ΪΖζ±ΫΜρ»ΪΖζΝΣ±ΫΈΣΝ§Ϋ”ΫαΙΙ,ΝΔΧεΉ®“Μ–‘ΒΊ¥°ΝΣΤπi,i+4ΈΜΒΡέœΜυ≤ύΝ¥¥”Εχ–Έ≥…Νρ¥ζΖζ±Ϋ«≈ΜΖΓΘ”Π”Ο¥ΥΖΫΖ®Ϋχ––ΝΥHIV-1≤ΓΕΨ“¬Ω«CAIΒΑΑΉΤ§ΕΈΒΡΗΡΙΙ…ηΦΤ,ΗΟΖΫΖ®ΒΟΒΫΒΡΕ© ικΡΡή”––ß“÷÷ΤHIV-1≤ΓΕΨΒΡ–Έ≥…,«“‘ΎΫαΚœΡήΝΠΓΔ¥©ΡΛΡήΝΠ“‘ΦΑΟΗΫβΈ»Ε®–‘…œΕΦ”≈”ΎΈ¥Ψ≠ΙΐΗΡ‘λΒΡ‘≠–ΈκΡ(ΆΦ7)ΓΘΗΟΖΫΖ®‘Ύ“ΜΕ®≥ΧΕ»…œ «Ε‘œ©ΧΰΗ¥Ζ÷ΫβΖ®ΒΡ≤Ι≥δ,«“‘ΎΖ¥”ΠΒΡΙΐ≥Χ÷––η“ΐ»κΫπ τ¥ΏΜ·ΦΝ, Ά§ ±“≤≤Μ–η“ΣΖ«≥ΘΑΚΙσΒΡΖ«Χλ»Μœ©ΜυΑ±ΜυΥα, “ρΕχΚœ≥…≥…±Ψ“≤¥σΈΣΫΒΒΆΓΘ
2.2.3 Νρ¥ζΥΡύΚ«≈ΜΖ
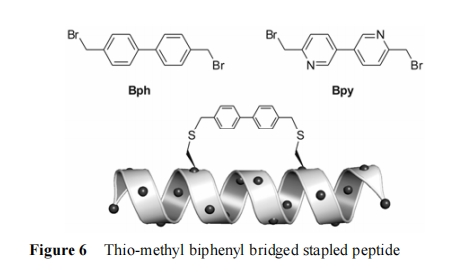
Brown Β»[123]‘ΎΕ‘ΑκκΉΑ±Υα≤–Μυ≤ύΝ¥ΗΡ‘λ ±, Ά®Ιΐ 3,6-Ε଻-1,2,4,5-ΥΡύΚ”κέœΜυΖΔ…ζ»Γ¥ζΖ¥”ΠΚσ–Έ≥…Νρ¥ζΥΡύΚΜΖά¥Έ»Ε® ΠΝ-¬ί–ΐΙΙœσΓΘΝρ¥ζΥΡύΚ«≈ΜΖ‘ΎΙβΜ·―ßΖ¥”Πœ¬Ω…ΖΔ…ζΝ―Ϋβ–Έ≥…Νρ«ηΥαθΞ≤ύΝ¥, ÷°ΚσΩ…‘ΎΑκκΉΑ±ΥαΒΡΉς”Οœ¬Η¥‘≠≥…‘≠κΡ(ΆΦ 8)ΓΘ
2.2.4 έœΜυ−œ©ΒψΜςΖ¥”Π
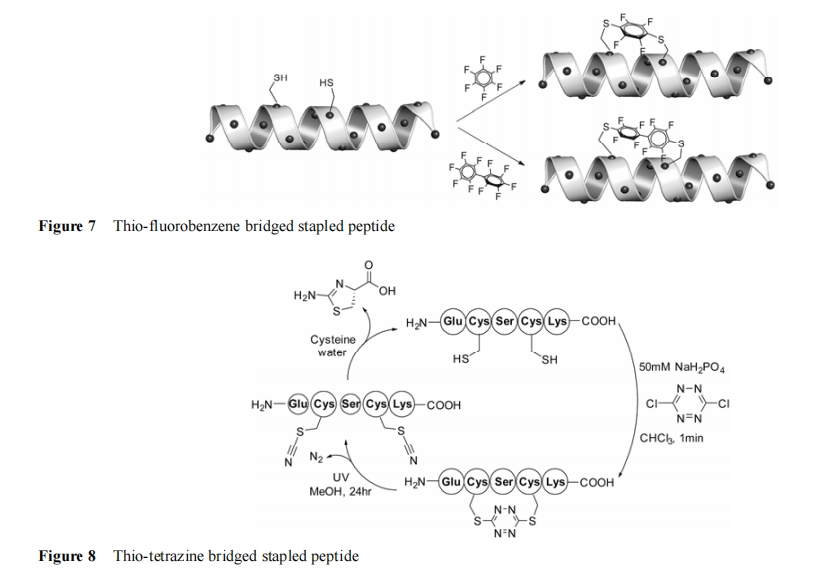
Wang Β»[124]±®ΒάΝΥάϊ”ΟέœΜυ−œ©ΒψΜςΖ¥”ΠΙΙΫ®Ε© ικΡ“‘ΦΑΕύκΡΒΡ¥σΜΖΜ·ΓΘ“‘≤ΜΆ§≥ΛΕ»ΒΡΕΰœ©”κΝΫΗωκΡΝ¥έœΜυ‘Ύ 365 nm Ιβ’’œ¬ “Έ¬Ζ¥”Π, ‘Ύ i, i+4 ”κ i, i+7 ΈΜΒψ÷°ΦδΙΙΫ®Νρ¥ζ«≈ΜΖΒΡΆ§ ±, Ε‘ΤδΥϊΙΌΡήΆ≈”–ΦΪΚΟΒΡΡΆ ή–‘, “ρ¥ΥΩ…“‘‘ΎΑ±ΜυΥα≤ύΝ¥Έ¥Ψ≠±ΘΜΛΒΡΉ¥Χ§œ¬Ϋχ––Ζ¥”Π (ΆΦ 9)ΓΘΥφΚσΥϊ Ο«±»ΫœΝΥΆ®Ιΐ RCM Ζ¥”Π–Έ≥…ΒΡ»ΪΧΦ«βΝ¥ Axin Ε© ικΡ[60]”κ”Π”ΟέœΜυ−œ©ΒψΜςΖ¥”ΠΗΡΙΙΕ© ικΡΒΡ ΠΝ-¬ί–ΐ–‘, ΖΔœ÷ΝΫ’Ώ‘Ύ‘≤Εΰ…ΪΤΉ÷–ΨΏ”–ΦΗΚθ“Μ÷¬ΒΡ ΠΝ-¬ί–ΐ±»άΐΓΘΥφΚσ”÷ΖΔœ÷»ΪΧΦ«βΝ¥ΒΡ p53 Ε© ικΡάύΥΤΈο”κΝρ¥ζ«≈ΜΖΙΙΫ®ΒΡΕ© ικΡ‘Ύ“÷÷Τ p53-MDMX œύΜΞΉς”Ο ±±μœ÷≥ωΝΥœύΥΤΒΡ“÷÷ΤΜν–‘, ≤ΔΩ…―Γ‘ώ–‘ΒΡ”’ΒΦ p53 “Α…ζ–ΆœΗΑϊΒΡΒρΆωΓΘΗΟΖΫΖ®Ρή÷±Ϋ””Ο”ΎΈ¥±ΘΜΛΒΡΕύκΡ, Ά§ ±“≤≤Μ–η“ΣΫπ τ¥ΏΜ·ΦΝΒΡ Ι”Ο, Εχ«“’β÷÷άύΥΤ»ΪΧΦΝ¥ΒΡΝ¥Ϋ”±έΒΡ Ι”ΟΡήΙΜ±ήΟβ”κ¥σΧεΜΐΒΡΖΦœψΜυΆ≈ΖΔ…ζΖ«ΧΊ“λΒΡΖ¥”ΠΓΘ
2.2.5 έœΜυ−»≤ΒψΜςΖ¥”Π
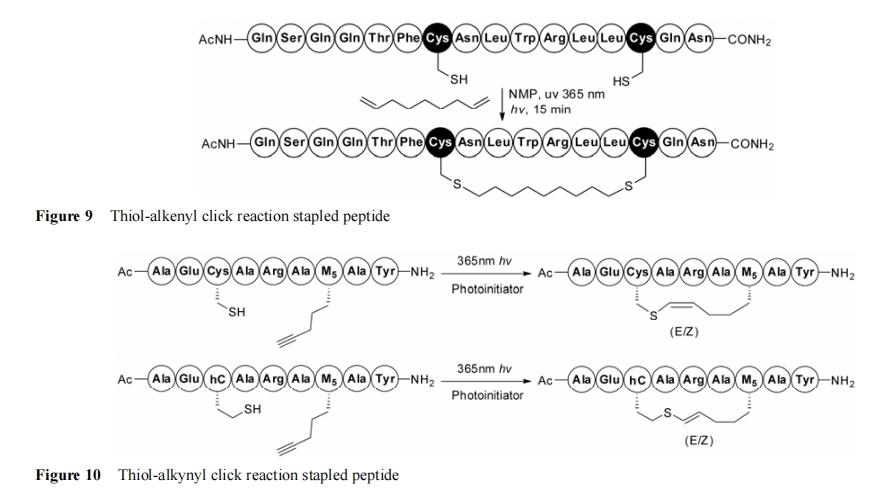
Tian Β»[125]±®ΒάΝΥ‘Υ”ΟΙβ“ΐΖΔΒΡΒΞΉιΖ÷έœΜυ−»≤ΒψΜςΖ¥”Π, ΗΏ–ßΦρΫύ‘Ύ≤ύΝ¥Έ¥Ψ≠±ΘΜΛΒΡ ΠΝ-¬ί–ΐκΡ…œΙΙΫ®Νρ−œ©«≈ΜΖΓΘ‘ΎκΡΝ¥ΒΡ i, i+4ΈΜ…œΖ÷±π“ΐ»κΈλ»≤Η Α±Υα (M5) ”κΑκκΉΑ±Υα, ΥφΚσ‘Ύ 365 nm ΒΡΙβ“ΐΖΔΧθΦΰœ¬Άξ≥…«≈ΜΖΒΡΙΙΫ®ΓΘ”ΟΗΟ≤Ώ¬‘Ε‘ΒςΫΎ¥ΤΦΛΥΊ ήΧε−Ι≤Ά§ΦΛΜνΦΝœύΜΞΉς”ΟΒΡ ΠΝ-¬ί–ΐκΡΫχ––ΗΡΙΙ, Ά®Ιΐ”κ»ΪΧΦ«βΝ¥ΗΡΙΙΚσΒΡΕ© ικΡΒΡ±»Ϋœ, œ‘ ΨΝρ−œ©«≈ΜΖΕ© ικΡ«Ή÷§–‘±δΜ·Ϋœ¥σΕχ«“œΗΑϊΡΛΕΨ–‘ΫœΒΆΓΘΝρΧΦΥΪΦϋΈΣΫχ“Μ≤ΫΒΡΙΠΡήΜ·ΧαΙ©ΝΥΩ… Ρή, Ά§ ±“≤ΖαΗΜΝΥΕ© ικΡΒΡΗΡΙΙΖΫΖ®, ΫΪΙ≈άœΒΡΝρ”κ»≤Ή‘”…ΜυΦ”≥…Ζ¥”Πά©’ΙΒΫΝΥΜ·―ß…ζΈο―ßΒΡ―–ΨΩ÷–ά¥(ΆΦ 10)ΓΘ
2.2.6 »≤ΧΰΗ¥Ζ÷ΫβΖ¥”Π
Cromm Β»[126]±®ΒάΝΥΫΪœ©ΧΰΗ¥Ζ÷Ϋβ (RCM) ”κ»≤ΧΰΗ¥Ζ÷Ϋβ (RCAM) Ζ¥”Π“‘’ΐΫΜΒΡΖΫ ΫΝΣΚœ”Π”Ο”ΎΕύκΡΒΡΫαΙΙΗΡ‘λ÷–, ΤδΙΊΦϋΜΖΫΎ‘Ύ”Ύ≥ΐΝΥœρΡΩ±ξκΡ÷–ΒΡΥΡΗωΜΖΚœΈΜΒψ (i, i+3, i+4, i+7) ÷–ΒΡΝΫ¥Π“ΐ»κœ©ΜυΑ±ΜυΥα (S5) “‘Άβ, ‘ΎΝμΆβΝΫΗωΈΜΒψ¥Π–η“ΐ»κ≤ύΝ¥ΈΣ»≤ΜυΒΡΖ«Χλ»ΜΑ±ΜυΥαΓΘ”…”ΎRCM Ζ¥”Π (ν…¥ΏΜ·) ΚΆ RCAM Ζ¥”Π (νβ¥ΏΜ·) ΧθΦΰΒΡ’ΐΫΜ–‘, ”κΥΪΕ© ικΡ[33]œύ±», Ω…“‘―Γ‘ώ–‘ΒΡΩΊ÷ΤΜΖΚœΒΡΒΡΈΜ÷Ο¥”Εχ Βœ÷Ε‘ΜΖκΡΆΊΤΥΫαΙΙΒΡΩΊ÷ΤΓΘΉς’ΏΫΪΗΟΖΫΖ®”Π”Ο”Ύ GTPase ΟΗ“÷÷ΤΦΝΒΡΜΖκΡΗΡ‘λ÷–≤ΔΒΟΒΫΝΥΜν–‘ΗΡ…ΤΒΡ“÷÷ΤΦΝ StRIP3 (ΆΦ 11)ΓΘ–η“Σ÷Η≥ωΒΡ «Υδ»ΜΉς’Ώ―Γ»ΓΒΡΗΡΙΙΜν–‘κΡœ»ΒΦΈο≤ΔΖ« ΠΝ-¬ί–ΐκΡ, ΒΪΗΟ―–ΨΩΉςΈΣΨ≠ΒδΕ© ικΡΖΫΖ®ΒΡ―”…λ÷ΒΒΟΫηΦχΓΘ

3 ΉήΫα”κ’ΙΆϊ
Ε© ικΡΆ®Ιΐ”Ο≤ύΝ¥ΜΖΚœΒΡΖΫΖ®ΧαΗΏΝΥ ΠΝ-¬ί–ΐκΡΒΡΙΙœσΈ»Ε®–‘¥”ΕχΦΪ¥σΒΡΧαΗΏ”κΑ–ΒΑΑΉΒΡ«ΉΚΆΝΠΓΔ¥ζ–ΜΈ»Ε®–‘ΓΔ¥©ΡΛ–‘ΓΘΕ© ικΡ‘Ύ“©–ßΓΔ≥…±Ψ“‘ΦΑΦΦ θΡ―Ε»…œΧν≤ΙΝΥ–ΓΖ÷Ή”“©Έο”κΒΞΩΥ¬ΓΩΙΧε‘ΎΒΑΑΉ−ΒΑΑΉœύΜΞΉς”ΟΈΣΑ–ΒψΒΡ“©Έο―–ΨΩ÷–ΗςΉ‘ΒΡ≤ΜΉψ, “―≥…ΈΣΜώΒΟ“‘ΒΑΑΉ−ΒΑΑΉœύΜΞΉς”ΟΈΣΑ–ΒψΒΡ“©Έοœ»ΒΦΈοΒΡ“Μ÷÷”––ßΆΨΨΕΓΘΆ®ΙΐΕ‘“―”–―–ΨΩΒΡΖ÷ΈωΩ…“‘ΖΔœ÷, ΡΩ«ΑΕ© ικΡΒΡ―–ΨΩ÷ς“ΣΦ·÷–‘ΎΝΫΗωΖΫœρ, Τδ“Μ «Ε‘Ζ÷Ή”…ζΈο―ßΚΆΫαΙΙ…ζΈο―ß―–ΨΩΖΔœ÷ΒΡΩ…ΒςΩΊΒΑΑΉ−ΒΑΑΉœύΜΞΉς”ΟΒΡΜν–‘ ΠΝ-¬ί–ΐκΡ≤…”ΟΨ≠ΒδΒΡœ©Μυ»ΪΧΦΕ© ι«≈≤Ώ¬‘ Β ©ΗΡ‘λ, ΗΟ―–ΨΩΖΫœρΤπ≤ΫΉν‘γ«“Φ¥ΫΪ–Έ≥… ΉΗωΕ© ικΡ–¬“©, ÷ΛΟςΝΥΕ© ικΡ“©ΈοΩΣΖΔΒΡΩ…–––‘; Νμ“ΜΗωΖΫœρ «‘ΎΚœ≥…ΖΫΖ®―ßΖΫΟφ≤…”Ο“―”–ΜρΩΣΖΔ–¬ΒΡ≈ΦΝΣΖ¥”Πά¥ΙΙΫ®ΨΏ”––¬ΫαΙΙΒΡ–¬–ΆΕ© ικΡ, ΗΟΖΫœρ“≤÷πΫΞ≥…ΈΣΝΥ”–ΜζΜ·―ß”κΜ·―ß…ζΈο―ßΒΡΫΜ≤φ―–ΨΩ»»ΒψΓΘΒΪ”…”ΎΜν–‘¬ί–ΐκΡΒΡΜώΒΟΖ«≥Θ“άάΒ”ΎΫαΙΙ…ζΈο―ßΒΡΨßΧεΫαΙΙΜρ «Ζ÷Ή”…ζΈο―ßΒΡΖΔœ÷, Ε‘”ΎΒΞΕά¥” ¬Κœ≥…ΖΫΖ®―ß―–ΨΩΒΡΩΈΧβΉιΕχ―‘ΆυΆυ≤…”Ο“―”–±®ΒάΒΡΜν–‘Ε© ικΡΉςΈΣœ»ΒΦΈοΜρ «≤…”ΟΈό…ζΈο―ßΜν–‘ΒΡΡΘ–Ά¬ί–ΐκΡΉςΈΣ―–ΨΩΕ‘œσΓΘΕ© ικΡ―–ΨΩ“―Ψ≠≥…ΈΣΫϋΡξά¥ΕύκΡ“©Έο―–ΨΩΒΡ»»Βψ, ’ΐ»γΕ© ικΡ―–ΨΩΝλ”ρΒΡΝλΒΦ’ΏWalensky ΫΧ ΎΥυ ω[127], ΗΟΖΫΖ®Ε‘ΕύκΡ–‘÷ ΒΡ–μΕύΗοΟϋ–‘ΗΡ…ΤΫΪ ΙΒΟΕύκΡ“©ΈοΒΡ―–ΨΩΒΟΒΫΗ¥–ΥΓΘΩ…“‘‘ΛΦϊ, ΥφΉ≈Ε© ικΡ―–ΨΩΒΡ≤ΜΕœ…ν»κ, ‘ΎΕ© ικΡΫαΙΙ…ηΦΤΓΔΚœ≥…ΖΫΖ®“‘ΦΑ–¬Α–ΒψΒΡΖΔœ÷ΖΫΟφΫΪ≤ΜΕœ”––¬ΒΡ―–ΨΩ≥…Ιϊ≤ζ…ζ, “≤±ΊΫΪΈΣΩΙ÷ΉΝωΓΔΩΙ≤ΓΕΨΓΔΩΙœΗΨζΗ–»ΨΓΔΡΎΖ÷ΟΎΒςΩΊΓΔ’οΕœ ‘ΦΝΒ»ΖΫΟφΒΡ“©Έο―–ΨΩΧαΙ©–¬ΒΡΥΦ¬ΖΚΆΫβΨωΖΫΑΗΓΘ
Οβ‘π…υΟςΘΚ±ΨΈΡΈΣ––“ΒΫΜΝς―ßœΑΘ§Αφ»®Ιι‘≠Ής’ΏΥυ”–Θ§»γ”–«÷»®Θ§Ω…ΝΣœΒ…Ψ≥ΐ
