ЖрыФХМСЊвЉЮяЃЈPeptide-drug conjugates, PDCЃЉЪЧМЬПЙЬхвЉЮяХМСЊЮяЃЈantibody-drug conjugates, ADCЃЉжЎКѓАаЯђжЮСЦЕФаТЗНЪНЁЃгыADCЯрБШЃЌPDCЕФКЫаФгХЪЦдкгкДЉЭИзщжЏФмСІЧПЁЂЛЏбЇКЯГЩИќШнвзЁЂЩњВњГЩБОИќЕЭЁЃФПЧАгаСНжжPDCвЉЮяБЛFDAХњзМгУгкжЮСЦАЉжЂЁЃPDCЕФжЮСЦаЇЙћЯджјЃЌЕЋзїЮЊАаЯђжЮСЦвЉЮяДцдкЮШЖЈадВюЁЂбЊвКбЛЗЪБМфЖЬЁЂбаЗЂЪБМфГЄЁЂСйДВПЊЗЂЙ§ГЬТ§ЕШЮЪЬтЁЃвђДЫЃЌСЫНтАЉЯИАћАаЯђPDCЕФзюаТбаОПНјеЙЁЂЮШЖЈадЮЪЬтЕФНтОіЗНАИЁЂМЦЫуЛњММЪѕИЈжњЦфбаЗЂЕФЗНАИвдМАЮДРДЕФЗЂеЙЗНЯђКмгаБивЊЁЃБОЮФДгPDCЕФНсЙЙКЭЙІФмГіЗЂЃЌДгАЉЯИАћАаЯђыФЃЈcancer cell-targeting peptideЃЌ CTPЃЉЕФбЁдёЁЂвЉДњЖЏСІбЇЬиадЁЂЮШЖЈадЕїПиЕШЗНУцЯЕЭГзлЪіСЫPDCsЕФзюаТбаОПНјеЙЃЌЯЃЭћФмЭЛГіЕБЧАДцдкЕФЮЪЬтКЭЮДРДЕФЗЂеЙЗНЯђЁЃ
1. ИХЪі
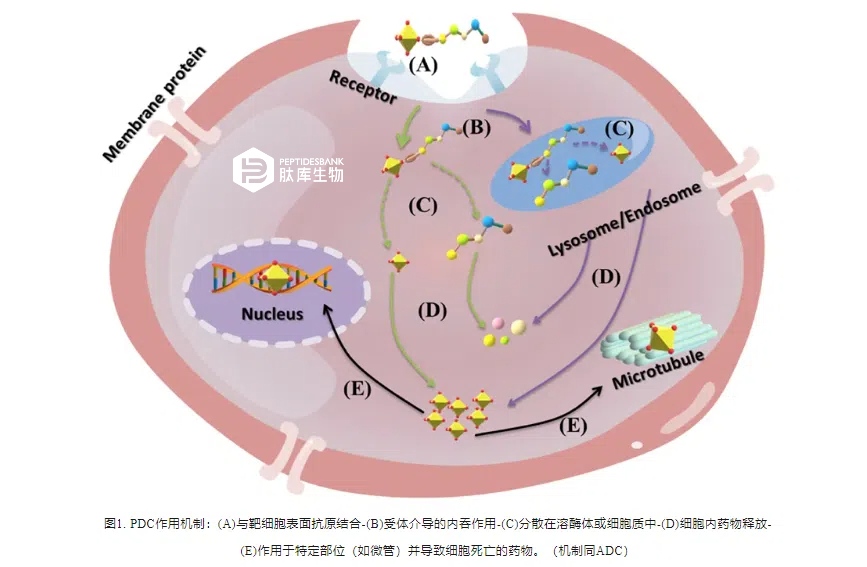
PDC гыADCЯрБШЃЌLinkersКЭDrugsВПЗжЯрЫЦЃЌВЛЭЌЕФЪЧPedtideКЭmAbЁЃCTPГЄЖШНЯЖЬЃЌПЩвдЬивьадЪЖБ№ВЂНсКЯЦфдкАЉЯИАћБэУцЬивьадБэДяЕФЪмЬхЃЌвВПЩвдв§ЕМвЉЮядкБэДяЪмЬхЕФАЉЯИАћБэУцаюЛ§ЃЌВЂЧвгЩгкЦфЬхЛ§НЯаЁЃЌИќШнвзЭЈЙ§АЉзщжЏЁЃОЁЙмСНепИХФюЯрЫЦЃЌЕЋНсЙЙКЭаджЪШДгаКмДѓВювьЃЌБэ1ЮЊPDC гыADCЖдБШНсЙћЁЃ
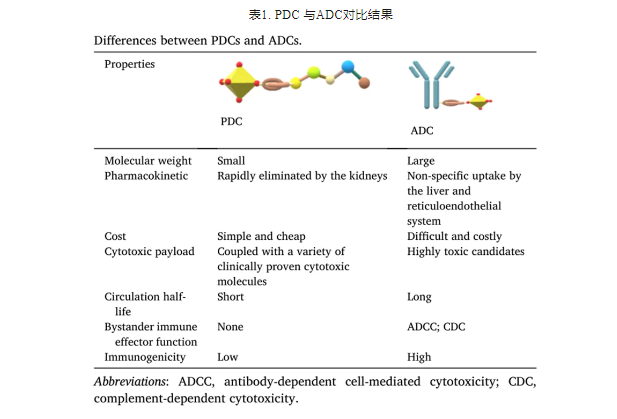
гЩгкADCКЭPDCЗжзгСПЕФВювьЃЌPDCОпгаНЯЧПЕФжзСіДЉЭИадЃЌУтвпдадНЯЕЭЁЃДЫЭтЃЌPDCКЭADCдкЬхФкЕФДњаЛЗНЪНВЛЭЌЁЃPDCЭЈЙ§ЩідрДњаЛЃЌЖјADCЭЈЙ§ИЮдрЁЃгЩгкPDCЕФыФађСаНЯЖЬЃЌЦфНсЙЙИќМгСщЛюЃЌетЪЙЕУв§ШыЗЧЬьШЛАБЛљЫсЃЌНјвЛВНаЮГЩЛЗзДыФЃЌвдМАНЋыФгыЦфЫћЗжзгЛЏбЇХМСЊИќШнвзЁЃЩЯЪівђЫидіЧПСЫPDCЕФАаЯђадЁЂЮШЖЈадКЭЭЈгУадЁЃ
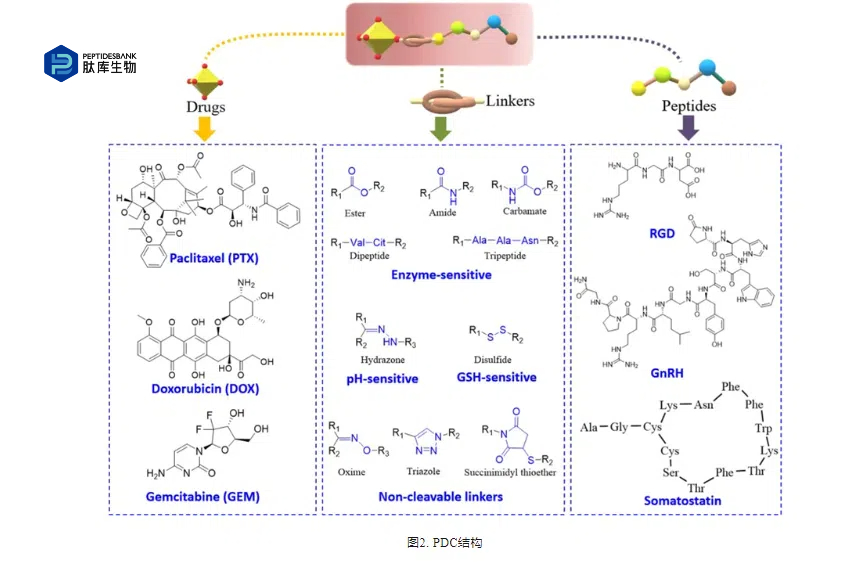
НќФъРДЃЌЫцзХЕААзжЪзщбЇЁЂЪЩОњЬхеЙЪОММЪѕЁЂmRNAеЙЪОММЪѕЁЂЙЬЯрыФКЯГЩЕШММЪѕПьЫйЗЂеЙЁЃдНРДдНЖрЕФаТЕФАаЯђыФБЛЗЂЯжЛђКЯРэЩшМЦЃЌетМЋДѓЕиДйНјСЫPDCЕФЗЂеЙЁЃдНРДдНЖрЕФPDCНјШыСйДВбаОПЃЌБэ2ЫљЪОЁЃ
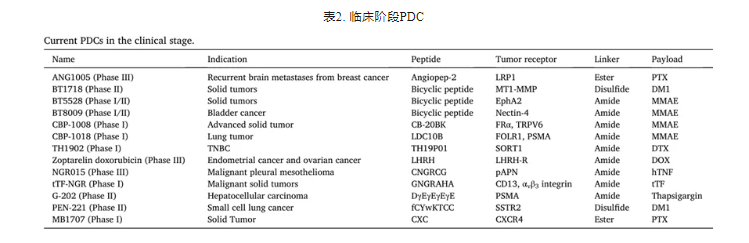
LutatheraКЭPepaxtoЪЧФПЧАЩЯЪаЕФPDCжЮСЦадвЉЮяЁЃLutatheraгЩХЕЛЊЙЋЫОПЊЗЂЃЌгк2018ФъЛёХњЃЌЪЙгУЩњГЄЫћЭЁРрвЉЮязїЮЊАаЯђыФЃЌЗХЩфадКЫЫи177 LuзїЮЊгааЇдиКЩЁЃЗХЩфадЭЌЮЛЫиПЩвдЭЈЙ§ЮяРэЗјЩфЩБЫРАЉЯИАћЁЃИУPDCзЂЩфЕНЛМепЬхФкКѓЃЌЭЈЙ§ЬивьадЪмЬхХфЬхЪЖБ№ВЖЛёжзСіЯИАћЃЌШЛКѓНЋЗХЩфадКЫЫив§ШыжзСізщжЏЃЌ177 LuЪЭЗХЕФИпФмІТЩфЯпзюжеЩБЫРжзСіЯИАћЁЃPepaxtoгЩOncopeptidesПЊЗЂЃЌгк2021ФъГѕЛёХњЃЌгЩвЛжжDNAЭщЛљЛЏМСЙВМлСЌНгЕНАаЯђАБЛљыФУИЕФыФЁЃгЩгкЦфИпЧзжЌадЃЌPepaxtoНјШыЯИАћКѓБЛАБЛљыФУИЫЎНтЃЌЪЭЗХГіЧзЫЎЕФDNAЭщЛљЛЏМСЃЌПЩв§Ц№жзСіЯИАћDNAЫ№ЩЫЃЌЩѕжСЕМжТЯИАћЫРЭіЁЃетжжPDCПЩгыЕиШћУзЫЩСЊКЯгУгкжЮСЦИДЗЂЛђФбжЮадЖрЗЂадЙЧЫшСіЕФГЩШЫЛМепЁЃВЛавЕФЪЧЃЌдк2021Фъ10дТ22ШеЃЌPepaxtoвђЦфIIIЦкСйДВЪЇАмЖјЭЫГіУРЙњЪаГЁЁЃЫфШЛЪаГЁЩЯЕФPDCБШADCЩйЃЌЕЋЛљгкЦфНсЙЙКЭадФмЕФВювьЃЌЮЊPDCЕФЮДРДЗЂеЙЬсЙЉСЫЮоЯоЕФПЩФмадЁЃБОЮФзлЪіСЫPDCЕФвЉДњЖЏСІбЇКЭЮШЖЈадЃЌВЂЖдЦфгІгУЧАОАНјааСЫеЙЭћЃЌВЂУшЪіСЫЛљгкИїжжАаЯђыФЕФPDCЕФзюаТбаОПНјеЙЁЃДЫЭтЃЌзмНсСЫМЦЫуЛњММЪѕЭЦЖЏPDCПЊЗЂЕФзюаТНјеЙЃЌШчЗжзгЖдНгЁЃВЂНщЩмСЫМИжжаТПЊЗЂЕФМЦЫуЛњЙЄОпдкPDCПЊЗЂжаЕФгІгУЁЃ
2. PDCЕФзюаТбаОПНјеЙ
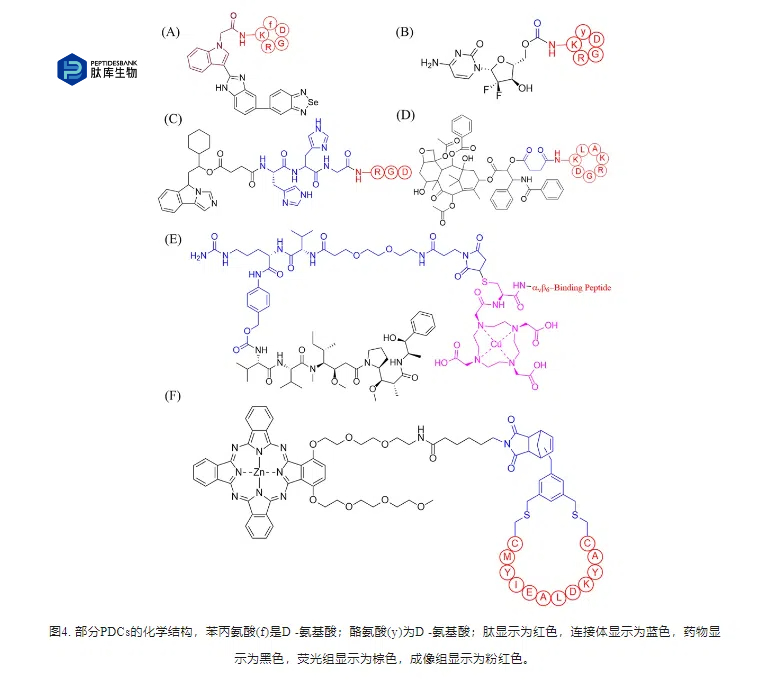
CTPЖдЯИАћЩЯЕФЬиЖЈЪмЬхОпгаСМКУЕФЧзКЭСІЃЌИУЪмЬхЕФЙ§БэДяЕМжТCTPДѓСПЙщГВЕНАЉЯИАћжаЃЌ вђДЫЃЌЭЈЙ§ЖржжВпТдЕФзщКЯЃЌCTPПЩвдГЩЮЊЕнЫЭвЉЮягааЇдиКЩЕФгааЇЙЄОпЃЌШчБэ3ЫљЪОЁЃ
2.1 АаЯђећКЯЫиЕФPDC
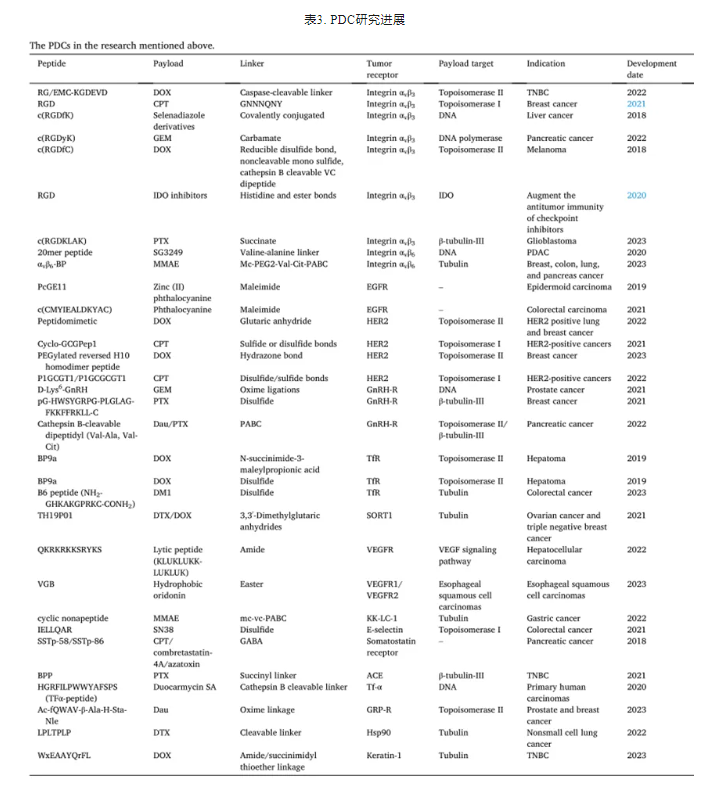
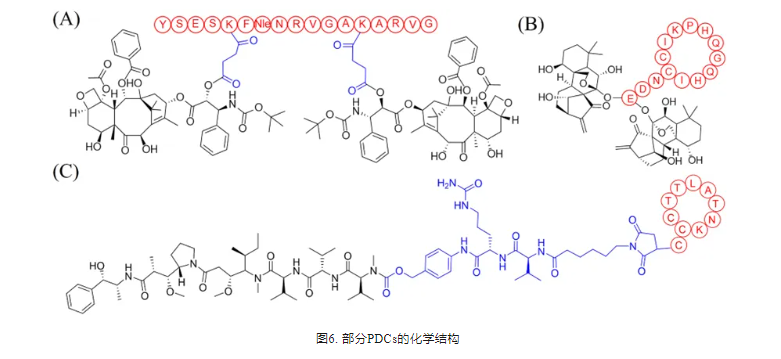
ећКЯЫиЪЧгЩІСКЭІТбЧЛљзщГЩЕФвьЖўОлЬхЪмЬхЃЌИКд№ЯИАћМфЁЂЯИАћФкКЭЯИАћЭтЛљжЪЕФеГИНЁЃЦљНёЮЊжЙЃЌвбМјЖЈГі18ЬѕІССДКЭ8ЬѕІТСДаЮГЩ24жжВЛЭЌЕФећКЯЫиЪмЬхЃЌІСvІТ3ећКЯЫиЪмЬхЕФЙ§БэДявбБЛжЄУїгыаэЖрАЉжЂгаЙиЁЃБШШчКкЩЋЫиСіЁЂНКжЪФИЯИАћСіКЭШщЯйАЉЁЃRGDыФМАЦфбмЩњЮягыІСvІТ3ећКЯЫиОпгаИпЖШЬивьадНсКЯЁЃБэУїЫќУЧгаЧБСІгУгкАаЯђАЉжЂжЮСЦЁЃRGDыФЙуЗКгУгкАаЯђИјвЉЁЃWangЕШШЫЪЙгУздзщзАађСаЃЈGNNNQNYЃЉНЋRGDгыЯВЪїМюЃЈCPTЃЉНсКЯЃЌаЮГЩСЫPDCЗжзгЃЌПЩвддкдЮЛздЗЂаЮГЩФЩУзЭХДиЁЃгЩгкRGDЕФАаЯђЙІФмЃЌCTSЕФжЮСЦаЇЙћЕУЕНдіЧПЃЌвЉЮяЕФзюДѓФЭЪмМССПвВДѓДѓЬсИпЃЌШчЭМ3ЫљЪОЁЃ
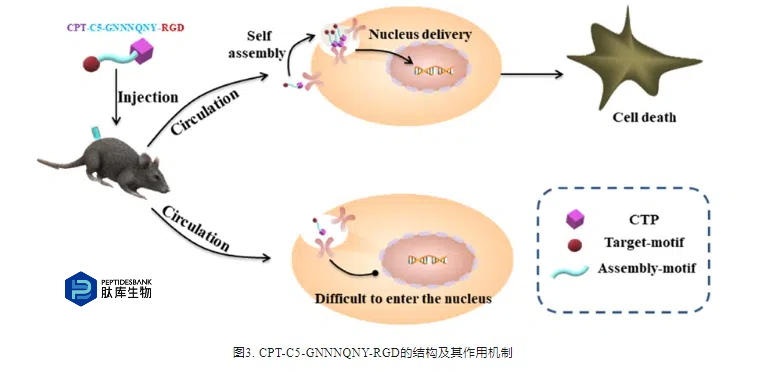
ZengЕШРћгУгЋЙтЛљЭХНЋc(RGDfK)гыЮјДњЖўпђбмЩњЮяЙВМлХМСЊЃЌЕУЕНЕФRGD-SeDдкЙ§БэДяІСvІТ3ећКЯЫиЕФHepG2ЯИАћжаБэЯжГігХвьЕФбЁдёадПЙАЉзїгУЁЃгЋЙтГЩЯёНсЙћНјвЛВНжЄЪЕИУPDCПЩбЁдёадЕнЫЭжСЙ§БэДяИУећКЯЫиЕФАЉЯИАћЃЈЭМ4AЃЉЁЃChatzisideriЕШШЫгУАБЛљМзЫсѕЅНЋGEMгыЛЗЛЏRGD (c(RGDyK)ХМСЊЃЈЭМ4BЃЉЃЌдкШЫбЊНЌжаБэЯжГіЮШЖЈадЃЌВЂж№НЅНЋGEMЪЭЗХЕНбЊвКжаЃЌдкWM266.4КЭA549ЯИАћжаЯдЪОГігХвьаЇЙћЁЃЮЊСЫНјвЛВНбаОПСЌНгЬхЕФДЬМЄЗДгІадЃЌLiangЕШВЩгУПЩЛЙдЖўСђМќЃЈSSЃЉЁЂВЛПЩЧаИюЕЅСђМќ(S)КЭзщжЏЕААзУИBПЩЧаИючгАБЫс-ЙЯАБЫсЖўыФЃЈVCЃЉзїЮЊСЌНгЛЗRGDЃЈc(RGDfC)ЃЉКЭDOXЕФСЌНгЮяЃЌЗжБ№ЩњГЩc(RGDfC)-SS-DOXЁЂc(RGDfC)-S-DOXКЭcЃЈRGDfCЃЉ(RGDfC)-VC-DOXЁЃЬхФкЯИАћЖОадЪдбщНсЙћЯдЪОЃЌc(RGDfC)-s-DOXКЭc(RGDfC)-vc-DOXЕФвЉаЇЪЧc(RGDfC)-ss-DOXКЭDOXЕФ1.4-2.0БЖЃЌетПЩФмгыc(RGDfC)-ss-DOXВњЩњDOX- shгаЙиЁЃГ§СЫЛЏСЦвЉЮяЭтЃЌPDCЛЙОГЃгУгкУтвпжЮСЦвЉЮяЕФПЊЗЂЁЃHanЕШШЫСЊКЯпХпсАЗ2,3-ЫЋМгбѕУИЃЈIDOЃЉвжжЦМСгыRGDЭЈЙ§зщАБЫсКЭѕЅМќаЮГЩЃЌаЮГЩЕФPDCПЩвдздзщзАЩњГЩФЩУзвЉЮяЁЃетаЉФЩУзвЉЮягааЇвжжЦжзСіжаЕФIDOЛюадЁЃдкЬхФкЯдЪОГіГжајвжжЦАЉЯИАћЩњГЄЃЌНЕЕЭШЋЩэЖОадЃЌВЂДѓДѓдіЧПГЬађадЯИАћЫРЭіХфЬхзшжЭМСЕФжЮСЦаЇЙћЃЈЭМ4CЃЉЁЃRizviЕШШЫПЊЗЂСЫвЛжжЫЋАаЯђыФЃЈc(RGDKLAK)ЃЉЃЌгЩRGDКЭАаЯђЯпСЃЬхЕФДйЕђЭіыФзщГЩЃЈKLAЃЉЃЌЭЈЙ§чњчъЫсХМСЊЕНPTXЃЈЭМ4DЃЉЁЃгыгЮРыPTXЯрБШЃЌаТаЭPDCОпгаИќКУЕФШмНтЖШКЭЯИАћЖОад(EC50= 8.32ЁР 0.09nM)ЃЌЬхФкЪЕбщНјвЛВНБэУїЃЌИУPDCЕФжзСіЩњГЄвжжЦзїгУдМЮЊPTXЕФ3БЖЁЃГ§СЫІСvІТ3ећКЯЫиЭтЃЌећКЯЫиІСvІТ6вВдкДѓЖрЪ§вШЯйЕМЙмЯйАЉжаЙ§БэДяЃЌвђДЫБЛШЯЮЊЪЧвЛИігаЯЃЭћЕФжЮСЦАаЕуЁЃЛљгкДЫЃЌMooreЕШШЫНЋЛљгкпСПЉБНЖўЕЊзПРрвЉЮяЕФгааЇдиКЩSG3249ХМСЊЕНвЛИіЬивьадНсКЯІСvІТ6ЕФ20ОлыФЩЯЃЌаЮГЩЙВщюSG3299ЁЃНсЙћБэУїЃЌАаЯђІСvІТ6ЕФPDC SG3299ЖдІСvІТ6ЕФЖОадИќДѓЃЌдкЬхЭтБэДяІСvІТ 6ЕФPDACЯИАћжъЃЈЮЊІСvІТ6вѕадPDACЯИАћжъЕФ78БЖЃЉЃЌдкЯрЭЌМССПЯТЃЌЦфЖОадУїЯдИпгкЗЧАаЯђPDACЯИАћжъЃЈИпДя15БЖЃЉЁЃетжжаТгБЕФPDCЮЊвШЯйАЉЬсЙЉСЫвЛжжаТЕФЬивьаджЮСЦбЁдёЁЃDavisЕШЭЈЙ§зщжЏЕААзУИПЩЧаИюСЌНгЬхНЋећКЯЫиІСvІТ6НсКЯыФКЭ64 CuИДКЯЮядгКЯзггыЯИАћЖОадвЉЮяMMAEНсКЯЃЈЭМ4EЃЉЃЌЛёЕУЕФ[64 Cu]-PDCОпгаНЯИпЕФШЫбЊЧхЮШЖЈадЁЂећКЯЫиІСvІТ6бЁдёадФкЛЏЁЂЯИАћЖОадвдМАСМКУЕФPETЯдЯёКЭЬхФквЉЖЏбЇЬиадЁЃ
2.2 АаЯђEGFRЕФPDC
БэЦЄЩњГЄвђзгЪмЬхЃЈEpidermal growth factor receptorЃЌEGFRЃЉЪЧвЛжждкЖржжАЉЯИАћжаЙ§БэДяЕФПчФЄЪмЬхЃЌЪЧжзСіжЮСЦЕФживЊАаЕуЁЃYuЕШЪЙгУЯпадEGFRНсКЯыФЃЈYHWYGYTPEVIЃЉзїЮЊжзСіАаЯђЛљЭХЃЌВЂгыЬЊнМХМСЊЁЃгыEGFRЕЭБэДяЕФШЫШщЯйАЉMCF7ЯИАћЯрБШЃЌетжжыФ-ЬЊнМХМСЊЮядкЙ§БэДяEGFRЕФБэЦЄбљАЉA431ЯИАћжаБэЯжГіИќИпЕФЯИАћЩуШЁКЭЖРЬиЕФЙтМЄЛюЯИАћЖОадЁЃДЫЭтЃЌChuЕШШЫНЋыФ-ЬЊнМХМСЊЮяжаЕФЯпадыФЬцЛЛЮЊЛЗзДEGFRНсКЯыФЃЈCMYIEALDKYACЃЉЃЌгЩгкЯпадыФЮШЖЈадНЯВюЃЌПЩФмгАЯьPDCЕФвЉДњЖЏСІбЇКЭЩњЮяРћгУЖШЁЃгыСНжжEGFRвѕадАЉЯИАћ(HeLaКЭHEK293)ЯрБШЃЌетжжаТЛёЕУЕФыФ-ЬЊнМХМСЊЮяБЛСНжжEGFRбєадАЉЯИАћжъЃЈHT29КЭHCT116ЃЉгХЯШЮќЪеЃЌВЂБэЯжГіИќИпЕФЙтЯИАћЖОадЃЈЭМ4FЃЉЁЃ
2.3 АаЯђHER2ЕФPDC
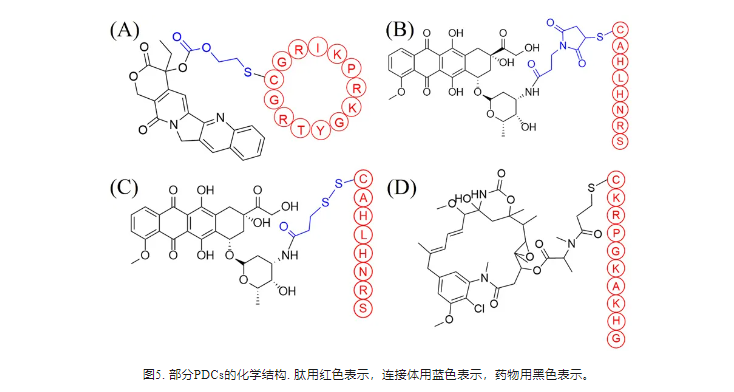
Г§СЫEGFRЃЌEGFRЕААзМвзхЕФСэвЛИіГЩдБЃЌHER2дкИїжжРраЭЕФАЉжЂжавВЙ§БэДяЃЌШчШщЯйАЉКЭЗЮАЉЁЃдкSonjuЕФбаОПжаЃЌHER2АаЯђыФгыDOXНсКЯЃЌЕУЕНЕФPDCБЛАќзАдкpHвРРЕЕФжЌжЪЬхвЉЮяЕнЫЭЯЕЭГжаЃЌгые§ГЃЩњРэЬѕМўЯрБШЃЌдкжзСіЮЂЛЗОГжаpHвРРЕЕФPDCЕнЫЭгааЇЃЌетДѓДѓдіЧПСЫPDCЕФЯИАћЩуШЁЁЃДЫЭтЃЌгыгЮРыDOXЯрБШЃЌЫћУЧЙлВьЕНАаЯђвЉЮяЕнЫЭдіМгСЫПЙжзСіЛюадЃЌЭЌЪБНЕЕЭСЫЖОадЁЃZhouЕШШЫЛљгкЧњЭзжщЕЅПЙ-HER2НсКЯФЃаЭЩшМЦСЫcyclog-gcgpep1ЃЌШЛКѓгыCPTХМСЊЁЃЙЙНЈЕФPDCдкSK-BR-3КЭNCI-N87ЯИАћжаОпгаНЯЧПЕФПЙдіжГЛюадЃЈЭМ5AЃЉЁЃLiuЕШЬсГіСЫвЛжжЛљгкHER2АаЯђыФЕФаТаЭPDCЃЌРћгУЫсУєИаЕФыъМќзїЮЊСЌНгМСЃЌПЩвдзМШЗЕиНЋDOXЪфЫЭЕНHER2бєадSKBR-3ЯИАћЃЌДгЖјдіЧППЙжзСіСЦаЇЃЌЭЌЪБНЕЕЭШЋЩэЖОадЁЃгыгЮРыDOXЯрБШЃЌИУPDCЕФЯИАћЩуШЁИп2.9БЖЃЌIC50НіЮЊгЮРыDOXЕФШ§ЗжжЎвЛЁЃДЫЭтЃЌЬхЭтЪЕбщБэУїЃЌPDCПЩЯджјвжжЦаЁЪѓHER2бєадШщЯйАЉвьжжвЦжВЮяЕФЩњГЄЁЃЮЊСЫдіЧПHER2АаЯђыФгыЪмЬхжЎМфЕФЧзКЭСІЃЌZhouЕШВЩгУМЦЫуЛњФЃФтЕФЗНЗЈЩшМЦСЫЫЋЬивьадШкКЯыФP1GCGT1КЭP1GCGCGT1ЃЌгыЕЅвЛАаЯђыФЯрБШЃЌетаЉаТыФгыHER2ЕФНсКЯФмСІЯджјдіЧПЃЌВЂЧвЖдHER2бєадЯИАћОпгаЬивьадАаЯђзїгУЃЌОCPTХМСЊЛёЕУЕФPDCОпгаИќЧПЕФПЙжзСіЛюадКЭИќКУЕФАВШЋадЁЃ
2.4 АаЯђGnRH-RЕФPDC
GnRHЪЧвЛжжгЩЯТЧ№ФдКЯГЩЕФЪЎыФЃЌЦфЪмЬхдкЖржжЪЕЬхжзСіжаЙ§БэДяЁЃVrettosЕШШЫГЩЙІНЋD-Lys6-GnRHгыGEMНсКЯЃЌЙЙНЈСЫЫсЯьгІаЭGnRH-RАаЯђPDCЃЈGOXG1ЃЉЁЃGOXG1ВЛНіЮШЖЈадИпЃЌЖјЧвБЛАЉЯИАћИпЖШЩуШЁЁЃвЉДњЖЏСІбЇЪЕбщНсЙћЯдЪОЃЌGOXG1ФмЯджјЬсИпGEMбЊвЉХЈЖШЁЃЮЊСЫНјвЛВНЬсИпвЉЮяЩуШЁаЇТЪЃЌDengЕШШЫНЋАаЯђGnRHЪмЬхЕФыФКЭЯИАћДЉЭИыФЃЈCPPЃЉгыPTXХМСЊЃЌЛёЕУЕФЯИАћДЉЭИPDCНЋPTXжЧФмЕнЫЭЕНЙ§БэДяGnRHЪмЬхЕФMCF-7ЯИАћжаЁЃPDCЕФЯИАћЩуШЁТЪЪЧгЮРыPTXЕФСНБЖЁЃДЫЭтЃЌЯИАћЖОаддіЧПЃЌIC50ЮЊ3.8 nM ЃЈPTX IC50ЮЊ6.6 nMЃЉЁЃвЉЮядкжзСіЯИАћФкЕФбЁдёадКЭПиЪЭНЋжБНггАЯьвЉЮяЕФСЦаЇЁЃ
2.5 АаЯђTfRЕФPDC
зЊЬњЕААзЪмЬхЃЈTfRЃЉЪЧвЛжжIIаЭПчФЄЬЧЕААзЃЌЭЈЙ§ЯрЛЅзїгУдкЯИАћЬњЩуШЁжаЦ№ЙиМќзїгУЁЃTfRдкжзСіЯИАћжаЙ§БэДяЃЌЕЋдке§ГЃзщжЏжае§ГЃБэДяЃЌЪЧжзСіжЮСЦЕФгааЇАаЕуЁЃLiЕШШЫРћгУN-чњчъѕЃбЧАЗ-3-ТэРДѕЃбЧАЗБћЫсзїЮЊСЌНгМСЃЌНЋTfRАаЯђыФBP9aХМСЊжСDOXЁЃгыгЮРыDOXВЛЭЌЃЌКЯГЩзККЯЮяЭЈЙ§Й§БэДяЕФTfRБЛHepG2ЯИАћбЁдёадЕиЮќЪеЁЃДЫЭтЃЌИУХМСЊЮяЖдHepG2ЯИАћБэЯжГіЬивьадЖОадЃЈЭМ5BЃЉЁЃDOXЭЈЙ§ЖўСђМќгыBP9aНсКЯЃЌВњЩњBP9a-SS-DOXЃЈЭМ5CЃЉЃЌB6ыФЃЈNH2-GHKAKGPRKC-CONH2ЃЉПЩАаЯђCRCЯИАћБэУцЕФTfRЁЃZhangЕШШЫПЊЗЂСЫвЛжжУћЮЊLWJ-M30ЕФыФ-вЉЮяХМСЊЮяЃЌЫќгЩDM1КЭыФB6зщГЩЃЈЭМ5DЃЉЁЃLWJ-M30ЭЈЙ§ЦЦЛЕЮЂЙмЕААзгеЕМЯИАћЕђЭіЃЌдкВЛгАЯьаЁЪѓЬхжиЕФЧщПіЯТвжжЦHCT116ЯИАћЕФЩњГЄЁЃ
2.6 АаЯђSORT1ЕФPDC
SORT1ЪЧвЛжждкЖржжЖёаджзСіжаБэДяЕФЖрЙІФмЕААзЁЃЛљгкSORT1ЪмЬхЃЌCharfiКЭCurrieЕШПЊЗЂСЫыФ-DTXХМСЊвЉЮяTH1902КЭыФ-DOXХМСЊвЉЮяTH1904ЁЃдкЬхЭтЃЌTH1902ЃЈЭМ6AЃЉЯдЪОЖдMDA-MB-231ЯИАћгаНЯЧПЕФПЙдіжГКЭПЙЧЈвЦзїгУЁЃдкЬхФкЃЌгыDTXЯрБШЃЌTH1902дкMDA-MB-231КЭHCC-70аЁЪѓвьжжвЦжВФЃаЭжаПЩЕМжТжзСіЯћЭЫЃЌЧвЮДв§Ц№жаадСЃЯИАћМѕЩйЁЃетаЉЪ§ОнБэУїTH1902ЭЈЙ§SORT1ЪмЬхНщЕМЕФЛњжЦЖдTNBCОпгаНЯИпЕФЬхФкгааЇадКЭАВШЋадЁЃ
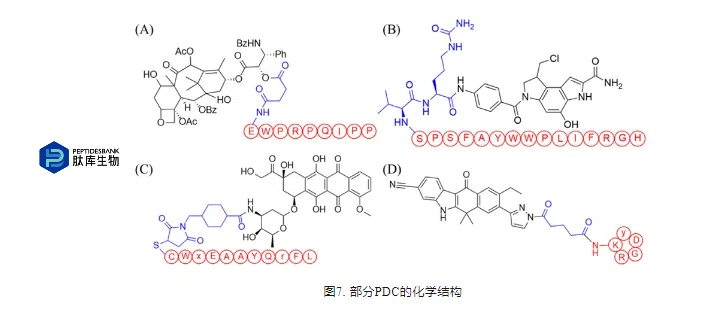
2.7 АаЯђVEGFRЕФPDC
дНРДдНЖрЕФжЄОнБэУїЃЌПЙбЊЙмЩњГЩЪЧвЛжжаТаЫЕФЁЂгаЧАЭОЕФжЮСЦАЉжЂЕФВпТдЁЃбЊЙмФкЦЄЩњГЄвђзгЃЈVascular endothelial growth factorЃЌVEGFЃЉМАЦфЪмЬхЃЈVEGFRЃЉЪЧжзСібЊЙмЩњГЩЕФЙиМќЗжзгЧ§ЖЏвђзгЃЌгШЦфЪЧVEGFR1КЭVEGFR2ВЮгыжзСіЯИАћКЭбЊЙмЯЕЭГЕФЕїПиЁЃWangЕШРћгУVEGFRАаЯђыФЃЈQKRKRKKSRYKSЃЉгыСбНтыФЃЈKLUKLUKKLUKLUKЃЉХМСЊЩњГЩPDCЃЌЗЂЯжPDCЖдИЮАЉЯИАћгаЧБдкЖОадЁЃQiЕШШЫНЋVEGFR1/VEGFR2ЫЋЬивьадыФVGB ЃЈCIKPHQGQHICNDEЃЉгыЪшЫЎЗжзгЖЋСшВнЫиЭЈЙ§ѕЅМќНсКЯЃЌЕУЕНPDCЃЈЭМ6BЃЉЃЌдкЪѕКѓжзСіИДЗЂФЃаЭжаЕФПЙжзСіСЦаЇЯджјдіЧПЁЃ
2.8 ЦфЫћАаЕуЕФPDC
ББОХжнЗЮАЉПЙд1
(KK-LC-1)ЃЌжЛдкМИжжРраЭЕФАЉжЂжаБэДяЃЌАќРЈЮИАЉЃЌетвтЮЖзХЫќПЩФмЪЧвЛИігаЯЃЭћЕФвЉЮяЪфЫЭАаЕуЁЃChenЕШШЫЪзДЮНЋKK-LC-1ШЗЖЈЮЊPDCЩшМЦЕФЧБдкАаЕуЃЌВЂзюжеШЗЖЈСЫKK-LC-1АаЯђPDC (1131-MMAE)ЃЈЭМ6CЃЉЁЃ1131-MMAEПЩБЛKK-LC-1бєадЮИАЉЯИАћгааЇФкЭЬЃЌВЂЭЈЙ§зшжЭG2/MЦкЯИАћжмЦкЪЭЗХвЉЮягеЕМЯИАћЕђЭіЁЃ
TNBCЪЧвЛжждЄКѓНЯВюЕФвьжЪадШщЯйАЉбЧаЭЁЃбЊЙмНєеХЫизЊЛЛУИЃЈACEЃЉдкTNBCЯИАћжавьЮЛБэДяЃЌЖјдкЪмЬхбєадЕФШщЯйАЉЯИАћЛђНЁПЕЩіЯИАћжаВЛБэДяЁЃдкДЫЛљДЁЩЯЃЌGuoЕШЩшМЦВЂКЯГЩСЫBPP-PTXЙВщюЮяЃЈЭМ7AЃЉЃЌЭЈЙ§НЋЛКМЄыФдіЧПыФЃЈBPPЃЉгычњчъѕЃСЌНгМСКЭPTXНсКЯЁЃгыгЮРыPTXЯрБШЃЌBPP-PTXЖдаЏДјMDA-MB-468жзСіЕФвьжжвЦжВДЦаладТуЪѓОпгаИќКУЕФжзСівжжЦзїгУЁЃДЫЭтЃЌBPP-PTXдкМѕЗЪКЭАзЯИАћМѕЩйЗНУцЖОадзїгУНЯаЁЁЃзлЩЯЫљЪіЃЌBPP-PTXЪЧжЮСЦACEбєадTNBCЕФвЛжжаТгБгааЇЕФЕЭЖОаджЮСЦЗНЗЈЁЃCartwrightЕШШЫЭЈЙ§ЙЬЯрКЯГЩЃЌЭЈЙ§зщжЏЕААзУИBПЩЧаИюСЌНгЮяНЋThomsen-FriedenreichАаЯђыФгыduocarmycin SAХМСЊЃЌЕУЕНЕФPDCЃЈЭМ7BЃЉбЁдёадвжжЦБэДяTf- ІСЕФжзСіЯИАћЁЃЮИУкЫиЪЭЗХыФЪмЬхЃЈGRP-RЃЉдкШщЯйАЉЁЂЧАСаЯйАЉЁЂвШЯйАЉКЭаЁЯИАћЗЮАЉЕШжзСізщжЏжаОљгаЙ§БэДяЃЌЪЧжзСіГЩЯёЁЂеяЖЯКЭжЮСЦЕФживЊАаЕуЁЃдкДЫЛљДЁЩЯЃЌGomenaЕШШЫПЊЗЂСЫвЛжжАаЯђGRP-RЕФPDCЃЌЯдЪОГіУїЯдЕФжзСіЯИАћПЙдіжГЛюадЁЃWxEAAYQrFLЪЧвЛИі10ОлЯпадНЧЕААз-1АаЯђыФНазі18-4ЁЃZiaeiКЭЭЌЪТЩшМЦСЫвЛжж18-4-DoxХМСЊЮяЃЈЭМ7CЃЉЃЌЖдTNBC MDA-MB-231ЯИАћОпгаИпЬивьадЖОадЁЃгыгУDOXЃЌНсКЯЮяДІРэаЁЪѓдкжзСіжаЛ§РлЕФDOXУїЯддіМгЃЈ7БЖЃЉЃЌжЮСЦаЇЙћИќКУЁЃ
3. PDCsЕФвЉДњЖЏСІбЇЬиеї
вЛАуРДЫЕЃЌPDCЕФДѓаЁКЭЗжзгСПНщгкаЁЗжзгвЉЮяКЭADCжЎМфЃЌетЪЙЕУPDCЕФЩјЭИадгХгкADCЃЌЕЋвВЕЭгкаЁЗжзгвЉЮяЃЛЖдPDCЕФЮќЪеВЛШчаЁЗжзгвЉЮяЁЃДЫЭтЃЌыФНјШыЬхФкКѓЃЌПЩвдЭЈЙ§ИїжжЗНЪНДгЬхФкХХГіЁЃгЩгкПкЧЛКЭЮИГІЕРжаКЌгааэЖрЕААзУИЃЌШчЕэЗлУИЁЂвШЕААзУИЁЂєШыФУИКЭУгФ§вШЕААзУИЃЌвђДЫPDCЭЈГЃЭЈЙ§зЂЩфЖјВЛЪЧПкЗўИјвЉЁЃДЫЭтЃЌЬьШЛЖрыФдкИјвЉКѓКмПьБЛбЊвКжаЕФЕААзУИЪЖБ№КЭЧхГ§ЁЃР§ШчЃЌЕквЛИіОХњзМЩЯЪаЕФPDC melflufenЃЌЮоТлЭЈЙ§жааФОВТіЕМЙмЛЙЪЧЭтжмОВТіЕМЙмИјвЉЃЌдкбЊНЌжаЖМгаЯрЫЦЕФЖЬБЉТЖЁЃвђДЫЃЌаэЖрбаОПепдкЬсИпЖрыФЕФЮШЖЈадзіГіСЫКмДѓЕФЙБЯзЁЃдкЩідржаЃЌгЩгкЫќУЧЕФЕЭЗжзгСПЃЌPDCЛсКмПьБЛЙ§ТЫЕєЃЌШЛКѓБЛЯћГ§ЃЌГ§СЫжБНгДњаЛЭтЃЌЯИАћФкЭЬзїгУКЭПьЫйАаЯђвЉЮяДІжУвВЪЧыФЯћГ§ЕФжївЊЭООЖЁЃPDCЕФЗжВМЪмЦфЗжзгСПЁЂЪшЫЎадКЭШмНтЖШЕФгАЯьЁЃРэЯыЧщПіЯТЃЌPDCгІЭЈЙ§ЖрыФгыЯИАћБэУцЪмЬхЕФЬивьадНсКЯЃЌбЁдёадЕиЗжВМдкжзСіВЁБфЧјгђЁЃPDCПЩвдЭЈЙ§АћЭЬзїгУКЭФкЛЏЕШПчФЄзїгУДгЯИАћЭтПеМфзЊвЦЕНЯИАћФкПеМфЁЃГ§СЫжзСіЧјгђЭтЃЌPDCЛЙЗжВМдкЩэЬхЕФЦфЫћзщжЏКЭЦїЙйжаЁЃР§ШчЃЌ177Lu-DOTATATEдкИјвЉКѓ4аЁЪБЗжВМгкЩідрЁЂИЮдрЁЂЦЂдрЁЂДЙЬхКЭМззДЯйжзСіВЁдюЁЃетжжЗжВМЬиеїжївЊЪЧгЩгкPDCЯрЖдгкаЏДјЕЅПЫТЁПЙЬхЕФADCЗжзгСПИќаЁЃЌЕМжТPDCгыПЙдЕФНсКЯСІБШЕЅПЫТЁПЙЬхШѕЃЌвђДЫPDCЕФзщжЏАаЯђадВЛШчADC, ДЫЭтЃЌгыADCВЛЭЌЃЌ177Lu-DOTATATEВЛБЛИЮдрДњаЛЁЃ177Lu-DOTATATEЕФЯћГ§жївЊЗЂЩњдкЩідрЃЌдкИјвЉ5аЁЪБФкРлМЦХХаЙ44%ЃЌ 58%дк24аЁЪБФкЃЌ65%дк48аЁЪБФкЁЃИљОн177Lu-DOTATATEЕФАыЫЅЦкМЦЫуЃЌдк14ЬьФкЃЌ99%вдЩЯЕФLu-DOTATATEЛсБЛЯћГ§ЁЃPDCЕФЗжВМгаЪБвВЪмЕНЖрыФКЭаЁЗжзгвЉЮяЕФЙЙЯѓЕФгАЯьЁЃReveretЕШбаОПСЫДЉЫѓЯИАћДЉЭИыФ(S-CPP)ЕФЗжВМЃЌгаСНжжВЛЭЌНсЙЙЃЌL-S-CPP КЭ D-S-CPPЁЃдкЩњЮяЬхжаЃЌЫћУЧЕФбаОПЯдЪОЃЌГ§СЫдкИЮдрКЭЦЂдржаЯдЪОПЩдЄВтЕФЛ§РлЭтЃЌИјвЉ1аЁЪБКѓЃЌаФдрКЭИЮдржаЕФDаЭХЈЖШИпгкLаЭЖдгГЬхЃЌДЫЭтЃЌИјвЉ5аЁЪБКѓЃЌбЊНЌКЭЩідржавВЗЂЯжСЫЯрЖдНЯИпЕФDаЭЫЎЦНЁЃ
4. ЬсИпPDCЕФЮШЖЈад
гыЖрыФвЛбљЃЌPDCзїЮЊАаЯђвЉЮяЕФжївЊШБЕуЪЧЦфЬхФкЮШЖЈадЕЭЁЂАыЫЅЦкЖЬЃЌвђЮЊЖрыФдкбЊвКжаБЛЕААзУИбИЫйНЕНтЃЌВЂБЛЩідрЧхГ§ЁЃЬиБ№ЪЧдкЪЕЬхжзСіЕФжЮСЦжаЃЌашвЊбгГЄPDCЕФбЛЗЃЌЪЙЦфФмЙЛГфЗжДЉЭИжзСізщжЏЁЃвђДЫЃЌШчКЮЬсИпPDCЕФЮШЖЈадКЭАыЫЅЦкЪЧбаОПЕФжиЕуЁЃ
4.1 ЛЗЛЏ
ЭЈЙ§ЛЗЛЏРДдіЧПыФЕФЬивьадЁЂАВШЋадКЭаЇСІЪЧЛЗыФЕФвЛИіЛљБОгХЪЦЁЃFDAХњзМЕФыФжагаШ§ЗжжЎЖўЪЧЛЗзДЕФЃЌетаЉЛЗзДыФдкЯжДњжЦвЉЙЄвЕжаЗЂЛгзХживЊзїгУЁЃЛЗЛЏДјРДЕФдМЪјПЩвдЪЙыФСДЕФЙЙЯѓИќМгЮШЖЈЃЌДгЖјЬсИпЦфгыАаЕААзЕФНсКЯЧзКЭСІЃЌМѕЩйЗЧЬивьадНсКЯЁЃЭЌЪБЃЌЛЗЛЏЯджјМѕЩйСЫПЩгУЙЙЯѓЕФЪ§СПКЭНјШыУИДпЛЏЮЛЕуЕФПЩФмадЃЌДгЖјдіМгСЫЕААзжЪзщбЇПЙадКЭАыЫЅЦкЁЃДЫЭтЃЌЛЗЛЏПЩвдЭЈЙ§гыИќДѓЕФЯрЛЅзїгУБэУцНсКЯРДИЩШХЕААзжЪЕФЯрЛЅзїгУЃЌДгЖјдіМгыФЕФЙІаЇЁЃзмЕФРДЫЕЃЌгыЯпадыФЯрБШЃЌЛЗыФВЛНіПЩвдИФЩЦыФСДЕФНсЙЙаджЪЃЌЛЙПЩвдгХЛЏЦфвЉДњЖЏСІбЇаджЪЃЌШчЮќЪеКЭФЄЭЈЭИадЁЃР§ШчЃЌЛЗзДRGDЃЈc(RGDyK)ЃЉБШЯпадRGDИќОпбЁдёадКЭЮШЖЈадЃЌБЛЙуЗКгУгкдіЧПгыећКЯЫиЕФНсКЯЁЃJH-VII-139-1ЪЧвЛжжЕЭФЩФІЖћSRPK1вжжЦМСЁЃLeonidisЕШШЫНЋJH-VII-139-1гыcЃЈRGDyKЃЉХМСЊЃЌЕУЕНЕФPDCЃЈЭМ7DЃЉЖдSRPK1МЄУИБЃГжжаЕШФЩФІЖћвжжЦЛюадЁЃ
4.2 аЮГЩФЩУзвЉЮяЪфЫЭЯЕЭГ
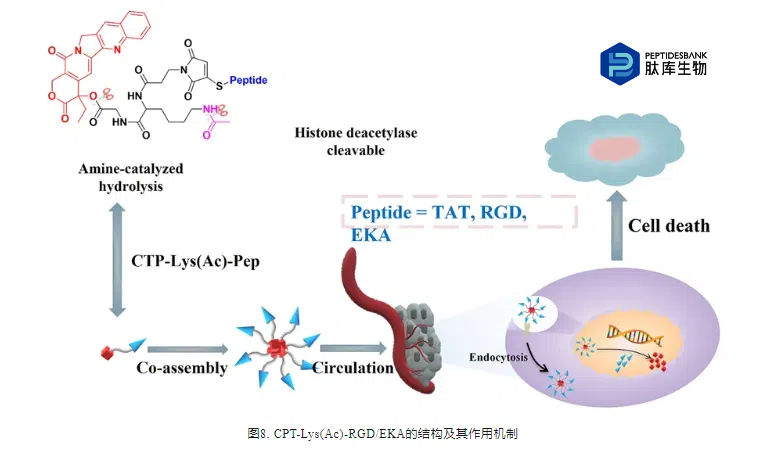
НќФъРДЃЌШЫУЧбаОПСЫФЩУзПХСЃРДЬсИпОлБНввЯЉЕФЮШЖЈадЁЃгЩгкФЩУзПХСЃОпгаРэЯыЕФЮяРэЛЏбЇаджЪЁЂАВШЋадЁЂЯрЖдШнвзКЯГЩКЭНЯГЄЕФАыЫЅЦкЃЌвђДЫПЩвдЬсИпPDCЕФећЬхЮШЖЈадЁЃР§ШчЃЌжЃЕШШЫКЯГЩСбНтыФPTP7КЭPTXжЦБИФЩУзСЃзгЃЈPPP NPs)ЁЃНсЙћБэУїЃЌPPP NPsЕФбЛЗАыЫЅЦкКЭХЈЖШ-ЪБМфЧњЯпЗжБ№БШгЮРыPTXГЄ56.5БЖКЭ7.5БЖЁЃДЫЭтЃЌдкИјвЉвЛЖЮЪБМфКѓЃЌгЮРыPTXжївЊЛ§ОлдкИЮдрЃЌЖјPPP NPsжївЊЗжВМдкЦЂдрЁЃЧзЫЎОлКЯЮяЕФв§ШыЪЙPDCОпгаСНЧзадЃЌЪЙЦфздОлМЏВЂаЮГЩФЩУзПХСЃЃЌДѓДѓЬсИпСЫPDCдкбЛЗжаЕФЮШЖЈадЁЃBaiЕШШЫЙЙНЈСЫCPT-Lys(Ac)-RGDКЭCPT-Lys(Ac)-EKAЙВщюЮяЃЌЫќУЧПЩвдздзщзА(ЭМ8)ЁЃ ЪЙгУEKAзїЮЊЯИАћЭтАаЯђШКЃЌдкШБЗІзщЕААзШЅввѕЃЛЏУИЕФЧщПіЯТЃЌЫќУЧЗЧГЃЮШЖЈЃЌЦфздзщзАПЩвдбгГЄдкбЊвКбЛЗжаЕФЪБМфЃЌВЂЭЈЙ§вжжЦжзСіЕФНјеЙКЭзЊвЦБэЯжГіСМКУЕФжзСіаюЛ§КЭНЯЧПЕФПЙжзСіЛюадЁЃ
4.3 ЬьШЛАБЛљЫсЕФаоЪЮ
вЛАуРДЫЕЃЌЬьШЛL-АБЛљЫсБЛДцдкгкбЊвККЭЬхФкЕФИїжжУИЫљЪЖБ№ЁЃвђДЫЃЌГ§СЫЛЗЛЏКЭФЩУзПХСЃЭтЃЌСэвЛжжГЃгУЕФЬсИпЖрыФЮШЖЈадЕФЗНЗЈЪЧаоЪЮЬьШЛАБЛљЫсЁЃаоИФыФѕЃАЗМќЩЯЕФNдзгЖдИФЩЦыФЕФвЉЮяаЮГЩКЭвЉДњЖЏСІбЇаджЪЦ№зХживЊзїгУЁЃЦфжаЃЌN-МзЛљЛЏвђЦфЭЈгУадКЭПЩБфаЮадЖјгІгУзюЮЊЙуЗКЁЃN-МзЛљЛЏПЩвдгааЇЕиаЮГЩгазщжЏЕФЧтМќЭјТчЃЌДгЖјЬсИпыФЕФЮШЖЈадЁЃдкФГаЉЧщПіЯТЃЌN-МзЛљЛЏЛЙПЩвдЬсИпАаыФЖдАаЕААзЕФЧзКЭСІЁЃДЫЭтЃЌЖдыФВрСДНјааТБЛЏвВПЩвдЬсИпЮШЖЈадЃЌЕЋетжжЗНЗЈШнвздіМгыФЕФЪшЫЎадЃЌЕМжТЦфОлМЏЖјЩЅЪЇЛюадЃЌвђДЫФПЧАЖдЦфбаОПНЯЩйЁЃбаОПШЫдБЛЙЬэМгСЫвЛжжЬиЪтЕФыФађСаРДБЃЛЄЫќВЛБЛНЕНтЁЃР§ШчЃЌYangЕШШЫдкыФRGDWRЕФNЖЫв§ШыІи -АБЛљБћЯЉЫсЃЌЕУЕНЕФІи RGDWRПЩвдБЃЛЄыФВЛБЛАБЛљыФУИЪЖБ№ЃЌДгЖјЬсИпЦфЮШЖЈадЁЃДЫЭтЃЌбЊаЁАхОлМЏЪдбщБэУїЃЌаоЪЮыФгывдЧАвЛбљгааЇЁЃЭЌбљЃЌLiuЕШШЫЖдPMAP-36PПЙОњыФЕФNЖЫШтЖЙЫсѕЅНјаааоЪЮЃЌЕУЕНСЫMyr-36PWЃЌгывдЭљЕФбаОПЯрБШЃЌMyr-36PWОпгаИќКУЕФПЙОњЛюадКЭИќИпЕФЮШЖЈадЁЃГ§СЫЩЯЪіАБЛљЫсЕФЛљБОаоЪЮЭтЃЌвЛаЉбаОПШЫдБЬсГіЪЙгУКЌгаN-ШЁДњИЪАБЫсЃЈN-GlysЃЉЕФРрыФРДКЯГЩыФбљЛЏКЯЮяЃЌгЩгкЦфЙЧМмВЛКЌѕЃАЗМќЃЌвђДЫОпгаИќКУЕФУИЮШЖЈадЁЃ
4.4 в§ШыЗЧЬьШЛАБЛљЫс
ЬьШЛЖрыФОпгаЮДОгХЛЏЕФНсЙЙЃЌЪЙЦфШнвзБЛУИНЕНтЛђЯћЛЏЁЃаэЖрбаОПБэУїЃЌВєШыЗЧЬьШЛАБЛљЫсПЩвдЬсИпЖрыФЕФЩњЮяЛюадКЭЕААзЫЎНтЮШЖЈадЁЃДЫЭтЃЌОнБЈЕРЃЌЗЧЬьШЛАБЛљЫсОпгаЬиЪтЕФНсЙЙКЭЮяРэЛЏбЇаджЪЃЈШчЗжзгЙЙЯѓШсШЭадЁЂНсЙЙЖрбљадЁЂТна§адКЭСНЧзадЃЉЁЃвђДЫЃЌКЌгаЗЧЬьШЛАБЛљЫсЕФыФЕФвЛМЖађСаПЩвдЮЊыФЬсЙЉСМКУЕФДњаЛЮШЖЈадКЭећЬхНсЙЙСщЛюадЁЃаэЖрбаОПШЫдБЖМЪдЭМгУD -АБЛљЫсШЁДњL -АБЛљЫсвдЬсИпЖрыФЕФУИЮШЖЈадЁЃ
5. МЦЫуЛњММЪѕИЈжњPDCЕФбажЦ
НќФъРДЃЌМЦЫуЛњММЪѕЗЂеЙбИЫйЃЌГЩБОВЛЖЯНЕЕЭЃЌОЋЖШВЛЖЯЬсИпЃЌгІгУЗЖЮЇВЛЖЯРЉДѓЃЌдкЩњЮявНбЇСьгђЕУЕНСЫЙуЗКЕФгІгУЁЃЛљгкЗжзгЖдНгЁЂЗжзгЖЏСІбЇФЃФтКЭШЫЙЄжЧФмЕФМЦЫуЛњММЪѕдкPDCЕФбаОПКЭПЊЗЂЕФИїИіЗНУцЗЂЛгзХдНРДдНживЊЕФзїгУЃЌШчзїгУЛњжЦЕФбаОПЁЂаТАаЕуЕФЗЂЯжЁЂыФЖЮКЭвЉЮяНсЙЙЕФдЄВтКЭгХЛЏЁЂЮќЪеЁЂЗжВМЁЂДњаЛЁЂХХаЙКЭЖОадЕФдЄВтЕШЃЈADMETЃЉЃЌВЂЖдЪЕбщЪ§ОнНјааЗжЮі(ЭМ9)ЁЃ
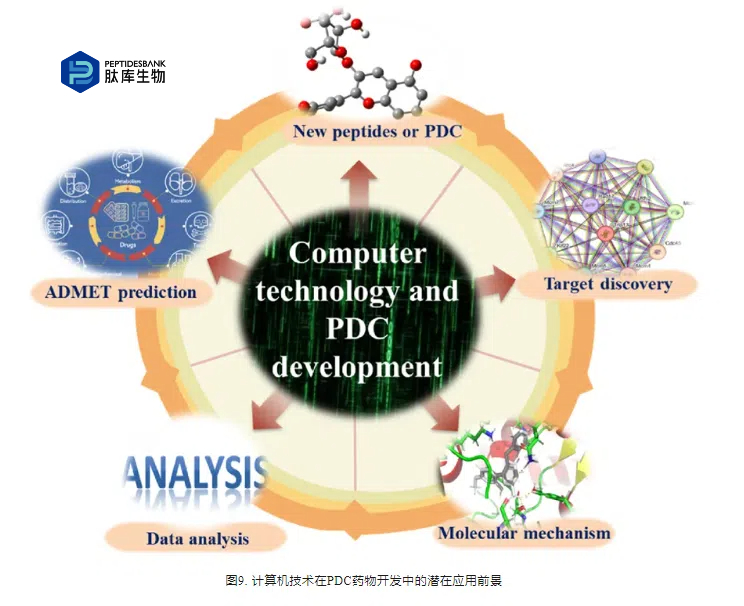
ЗжзгЖЏСІбЇФЃФтКЭЗжзгЖдНгПЩвдНвЪОЭЈЙ§баОПЗжзгМфЕФЯрЛЅзїгУРДбаОПвЉЮяЗжзггыАаЕААзЕФзїгУЛњжЦЃЌВЂНјвЛВНжИЕМвЉЮяЗжзгЕФЩшМЦЁЃParkЕШШЫЭЈЙ§НЋзщжЏЕААзУИBЬивьадыФЃЈRRЃЉгыЕЈжЫсЃЈBAЃЉХМСЊЩшМЦСЫвЛжжаТаЭPDC ЃЈRR-BAЃЉЁЃЫћУЧЪЙгУЗжзгЖЏСІбЇФЃФтРДНтЪЭRR-baгызщжЏЕААзУИBЕФЮШЖЈадгХгкгЮРыRRыФЁЃShokriЕШШЫЪЙгУыФ(ЬьЖЌАБЫс-ИЪАБЫс-ОЋАБЫс)ЃЌбЁдёадАаЯђжзСібЊЙмжаЙ§БэДяЕФАБЛљыФУИЃЌгыЗЧчоЬхПЙбзвЉЃЈнСЦеЩњЃЉНсКЯЪЙгУЁЃДЫЭтЃЌЭЈЙ§ЗжзгЖдНгжЄЪЕСЫPDCгыАБЛљыФУИЕФНсКЯБШгЮРынСЦеЩњИќЧПЁЃЭЈЙ§ЗжзгЖдНгЁЂЗжзгЖЏСІбЇФЃФтКЭШЫЙЄжЧФмЃЌПЩвддкДѓСПКђбЁЛЏКЯЮяжаПьЫйЗЂЯжгаЯЃЭћЕФвЉЮяЁЃДЫЭтЃЌЫќЛЙдЪаэбаОПШЫдБвдВЛЭЌЕФЗНЪННјааЕїећЃЌвдЬсИпвЉЮяЕФадФмЁЃZhouЕШШЫЛљгкПЙЬхгыHER2ЕФЗжзгЖдНгКЭНсКЯЗНЪНЃЌЩшМЦСЫвЛжжаТЕФЛЗзДыФGCGPep1ЁЃЪЕбщЪ§ОнжЄЪЕСЫИУЛЗыФЖдHER2ОпгаСМКУЕФЧзКЭСІЃЌВЂРћгУИУАаЯђыФГЩЙІЩшМЦВЂжЦБИСЫаТЕФАаЯђPDCЃЌМЦЫуЛњММЪѕПЩвдЭЈЙ§ШЫЙЄжЧФмЗжЮіДѓЙцФЃЕФЩњЮяаХЯЂбЇЪ§ОнЃЌЪЖБ№ЧБдкЕФМВВЁЯрЙиЛљвђЛђЕААзжЪзїЮЊЧБдкЕФPDCАаЕуЁЃ
РћгУЗжзгЖдНгКЭЗжзгЖЏСІбЇФЃФтЖдЗжзгНсЙЙНјаадЄВтКЭФЃФтЃЌгажњгкЗЂЯжЕААзжЪЕФНсЙЙЬиеїЃЌШЗЖЈПЩФмЕФНсКЯЮЛЕуКЭЯрЛЅзїгУЧјгђЁЃдквЉЮяЩшМЦжаЃЌШЫЙЄжЧФмКЭЦфЫћМЦЫуЛњММЪѕвВПЩвддкСйДВЧАдЄВтЫќУЧЕФADMETЬиадЁЃ
6. НсТлгыеЙЭћ
дкАЉжЂЕФжЮСЦжаЃЌДЋЭГЕФЛЏСЦгЩгкИБзїгУДѓЃЌвбОВЛФмТњзуЛМепЕФашвЊЃЌгыДЋЭГЛЏСЦвЉЮяЯрБШЃЌPDCзїЮЊвЛжжОЋзМЕФжзСіАаЯђжЮСЦЃЌРЉДѓСЫжЮСЦДАПкЃЌЬсИпСЫжЮСЦаЇЙћЃЌИФБфСЫШЫУЧЖдАЉжЂвЉЮяжЮСЦЕФШЯЪЖЁЃЕЋЪЧЃЌPDCвВДцдкЮШЖЈадВюЁЂАыЫЅЦкЖЬЕШЮЪЬтЁЃЫфШЛФПЧАЛЙУЛгаЭъУРЕФНтОіЗНАИЃЌЕЋЯраХPDCНЋКмПьИќКУЕигІгУгкСйДВЪЕМљЁЃ
вђДЫЃЌPDCАаЯђИјвЉЯЕЭГСьгђШдгаЮоЯоЕФПЩФмадЃЌашвЊдкЮДРДМИФъНјаабаОПЁЃЮДРДЕФЧїЪЦКЭбаОПШШЕуПЩФмАќРЈвдЯТМИИіЗНУцЁЃ
a. ЛљгкШЫдДЛЏПЙЬхЕФPDCЕФПЊЗЂЃКздЕЅПЫТЁПЙЬхГіЯжвдРДЃЌзюГЃгУЕФаЁЪѓЕЅПЫТЁПЙЬхдкСйДВеяЖЯКЭжЮСЦжаЗЂЛгСЫОоДѓЕФзїгУЁЃШЛЖјЃЌГЃгУЕФаЁЪѓЕЅПЫТЁПЙЬхОпгаНЯЧПЕФУтвпдадЁЃвђДЫЃЌЛљгкШЫдДЛЏПЙЬхЕФЖрыФдкПЊЗЂАВШЋгааЇЕФPDCжЮСЦвЉЮяжаОпгаживЊвтвхЁЃ
b.PDCЩњЮяЮШЖЈадЬсИпЃКгыаЁФІЖћвЉЮяЯрБШЃЌАаЯђPDCЬивьадЧПЃЌЕЋЩњЮяЮШЖЈадВюЁЃЮЊСЫЬсИпPDCдкЬхФкЕФЮШЖЈадЃЌШЗБЃЦфСЦаЇЃЌФПЧАе§дкбаОПЖдАаЯђыФКЭСЌНгЬхЕФаоЪЮЁЃФПЧАЃЌИїжжОпгаЬиЖЈЙІФмЕФФЩУзвЉЮяДЋЕнЯЕЭГвбБЛГЩЙІПЊЗЂЃЌВЂдкжзСіжЮСЦСьгђЗЂЛгСЫОоДѓЕФзїгУЁЃвђДЫЃЌНсКЯPDCФЩУзИјвЉЯЕЭГПЊЗЂСЫвЛжжаТаЭЕФАаЯђПЙЬхЛђАаЯђФЩУзыФЕФЖрЙІФмвЉЮяЁЃ
c. PDCгыМЦЫуЛњИЈжњвЉЮяЩшМЦЕФНсКЯЃКГ§СЫжївЊвРППЩњЮяЁЂЛЏбЇЕШММЪѕЃЌдкКмДѓГЬЖШЩЯПЩвдгыМЦЫуЛњИЈжњвЉЮяЩшМЦЗНЗЈЯрНсКЯПЊЗЂЁЃЭЈЙ§МЦЫуЛњЗжЮіЃЌЮвУЧПЩвдИќЖрЕиСЫНтыФМАЦфХфЬхжЎМфЕФЯрЛЅзїгУЁЃЮЊСЫСЫНтФПБъыФЪЧШчКЮЩшМЦЕФвдМАЫцКѓЕФPDCЪЧШчКЮПЊЗЂЕФЃЌЖдСЌНгЗНЗЈЁЂЗжзгЖЏСІбЇФЃФтКЭШЫЙЄжЧФмЪЙгУЬиЖЈЕФЫуЗЈРДдЄВтЁЂМЦЫуКЭЦРЙРетжжНсКЯЪмЬхгыХфЬхжЎМфЕФЧзКЭСІЁЃДЫЭтЃЌЮвУЧПЩвдЮЊФПБъЕААзЩшМЦаТЕФыФЃЌЛђепЭЈЙ§МЦЫуЗНЗЈдЄВтФПБъЕААзЛђЕААзжЪжаФГИіыФПЩФмНсКЯЕФЮЛжУЁЃ
d. PDCгыЖржжЪмЬхЛђПЙдНсКЯЃКвбЩЯЪаЕФPDCЛђе§дкПЊЗЂЕФPDCжаЪЙгУЕФАаЯђыФжївЊгыЕЅвЛЪмЬхНсКЯЁЃШчЙћЫљЪЙгУЕФЖрыФФмЙЛНсКЯЭЌвЛПЙдЕФВЛЭЌЮЛЕуЛђЭЌвЛАЉЯИАћБэУцЕФВЛЭЌЙ§БэДяЪмЬхЃЌдђЛсИФЩЦЪмЬхОлМЏЃЌМгЫйХМСЊвЉЮяЕФПьЫйФкЛЏЁЃ
e. ЫЋжидивЉPDCЕФЩшМЦЃКОЕфЕФыФХМСЊвЉЮяАќРЈыФЁЂСЌНгМСКЭгаЖОвЉЮяЁЃвђДЫЃЌPDCЕЅвЉв§Ц№ЕФФЭвЉЮЪЬтЪЧвЛИіВЛПЩЛиБмЕФЮЪЬтЁЃЫцзХвЉЮяжЦБИММЪѕЕФбИЫйЗЂеЙЃЌЫЋживЉЮяИКдиЪЙгУСНжжОпгаВЛЭЌзїгУЛњжЦЕФЯИАћжЦМСзїЮЊгааЇдиКЩЕФPDCгаЭћПЫЗўЪЙгУЕЅвЛвЉЮяЪБЕФЧБдкФЭвЉадЮЪЬтЁЃ
злЩЯЫљЪіЃЌЫцзХPDCАаЯђИјвЉЯЕЭГбаОПЕФЩюШыЃЌАаЯђадЧПЁЂЩњЮяЯрШнадКУЁЂАВШЋадИпЕФPDCНЋЕУЕНИќЙуЗКЕФПЊЗЂКЭгІгУЃЌЮЊШЫРрЕФГжајНЁПЕЗЂеЙзіГіживЊЙБЯзЁЃ
