2 增强多肽分子的药代稳定性
多肽的基本组成单元是氨基酸, 其本质与蛋白质相同, 因而多肽类分子是许多蛋白酶水解的底物, 而这一特点严重限制了多肽类药物的开发研究。一般而言, 大部分多肽类药物无法口服, 否则就会被胃蛋白酶以及胰蛋白酶等消化破坏; 其次, 即使通过注射给药, 多肽类药物也有可能在血液以及组织中被蛋白酶降解失活, 因此多肽类药物的生物利用度很低, 以至于多肽类分子在临床治疗中受到很大限制[23]。为了减弱或避免蛋白酶对多肽类分子的降解, 必须要利用化学方法或其他方法对多肽分子进行修饰改造, 以提高多肽的代谢稳定性, 为新药研发中解决多肽的代谢稳定性问题提供一些思路和参考。增强多肽分子代谢稳定性的主要方法包括非天然氨基酸修饰、伪肽化策略、逆肽策略、环化策略以及高级脂肪酸修饰、蛋白融合策略、聚乙二醇修饰等。
2.1 肽链骨架改造
对肽链骨架进行修饰和改造以增强多肽分子代谢稳定性的主要方法包括非天然氨基酸修饰、伪肽化策略、逆肽策略、环化策略等。
2.1.1 非天然氨基酸修饰
天然活性肽的组成常常都是天然氨基酸。天然活性肽容易受到体内蛋白酶降解, 从而降低其在体内的半衰期, 导致天然活性肽在体内发挥药效时间缩短, 不利于成药。β氨基酸作为非天然氨基酸, 在体内不易被蛋白酶识别水解, 在活性肽的结构改造与修饰中发挥重要作用。
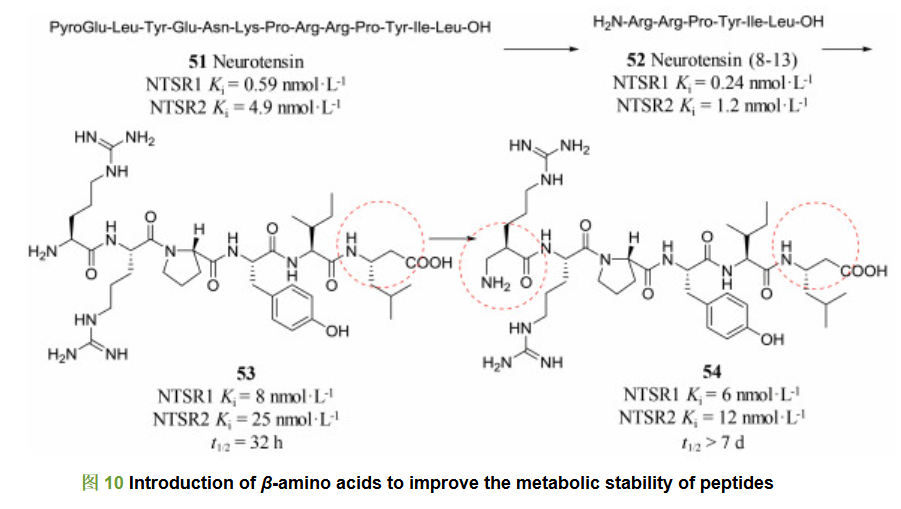
化合物51是一个神经降压素, 其作用于神经降压素受体1和2 (NTSR1和NTSR2)两个亚型。神经降压素及其受体与痛觉缺失的调节、食物摄取以及肿瘤生长具有密切关系[24]。研究人员对神经降压素51进行结构优化, 经过截断策略得到了含有第八位到第十三位氨基酸序列的简化肽52 (NTSR1 Ki=0.24 nmol・L-1; NTSR2 Ki=1.2 nmol・L-1), 其对NTSR1和NTSR2受体的活性均比神经降压素51有所提高。然而, 简化肽52容易受到体内酶代谢作用, 因此其在体内半衰期很短。针对这一特点, 研究人员尝试引入β氨基酸(图 10), 得到活性肽53, 其对NTSR1和NTSR2受体的活性虽然下降(NTSR1 Ki =8 nmol・L-1; NTSR2 Ki=25 nmol・L-1), 但半衰期延长至32 h。之后研究人员又将N端的精氨酸替换为β-精氨酸得到肽54。54相比于53活性略有提高(NTSR1 Ki =6 nmol・L-1; NTSR2 Ki=12 nmol・L-1), 而且54的半衰期大于7天[25, 26], 极大地提高了活性肽在体内的停留时间, 增强了活性肽在体内的药代稳定性。
 肽类小分子55是一个广泛研究的金属蛋白酶EP24.15 (endopeptidase)抑制剂。EP24.15与下丘脑对垂体功能的调节及血压调节有重要关联, 文献报道EP24.15还可能与Aβ蛋白的聚集和阿尔兹海默症(Alzheimer's disease, AD)相关, 因此EP24.15是精神系统疾病的研究热点。虽然肽类小分子55对EP24.15的抑制活性很强(IC50=0.06 μmol・L-1), 但它容易受到与EP24.15相关的蛋白酶――中性内肽酶EP24.11水解。因此, 研究人员的主要研发目标是提高55对中性内肽酶EP24.11的稳定性。他们尝试将55中的丙氨酸、酪氨酸和羧基末端分别用β-丙氨酸、β-苯丙氨酸和β-氨基丙酸替换, 得到β肽56对EP24.15的抑制活性虽然有所下降(IC50=2.8 μmol・L-1), 但对中性内肽酶EP24.11的稳定性显著提高(图 11), 几乎不受其降解影响[27]。
肽类小分子55是一个广泛研究的金属蛋白酶EP24.15 (endopeptidase)抑制剂。EP24.15与下丘脑对垂体功能的调节及血压调节有重要关联, 文献报道EP24.15还可能与Aβ蛋白的聚集和阿尔兹海默症(Alzheimer's disease, AD)相关, 因此EP24.15是精神系统疾病的研究热点。虽然肽类小分子55对EP24.15的抑制活性很强(IC50=0.06 μmol・L-1), 但它容易受到与EP24.15相关的蛋白酶――中性内肽酶EP24.11水解。因此, 研究人员的主要研发目标是提高55对中性内肽酶EP24.11的稳定性。他们尝试将55中的丙氨酸、酪氨酸和羧基末端分别用β-丙氨酸、β-苯丙氨酸和β-氨基丙酸替换, 得到β肽56对EP24.15的抑制活性虽然有所下降(IC50=2.8 μmol・L-1), 但对中性内肽酶EP24.11的稳定性显著提高(图 11), 几乎不受其降解影响[27]。
 研究人员用图 12的示意图解释引入β氨基酸可以提高肽类分子对中性内肽酶的稳定性。对于天然α多肽, 在特异性的蛋白酶切割位点, 水分子首先与酰胺键形成氢键作用, 从而有利于水分子对酰胺键的进攻最后完成酰胺键的切割; 对β多肽而言, 由于增加了一个亚甲基, 多肽整体的构象发生变化, 原本蛋白酶切割中心的水分子无法与酰胺键形成氢键, 不利于蛋白酶对酰胺键的切割, 因而β多肽比α多肽具有更强的抗水解能力[28]。
研究人员用图 12的示意图解释引入β氨基酸可以提高肽类分子对中性内肽酶的稳定性。对于天然α多肽, 在特异性的蛋白酶切割位点, 水分子首先与酰胺键形成氢键作用, 从而有利于水分子对酰胺键的进攻最后完成酰胺键的切割; 对β多肽而言, 由于增加了一个亚甲基, 多肽整体的构象发生变化, 原本蛋白酶切割中心的水分子无法与酰胺键形成氢键, 不利于蛋白酶对酰胺键的切割, 因而β多肽比α多肽具有更强的抗水解能力[28]。

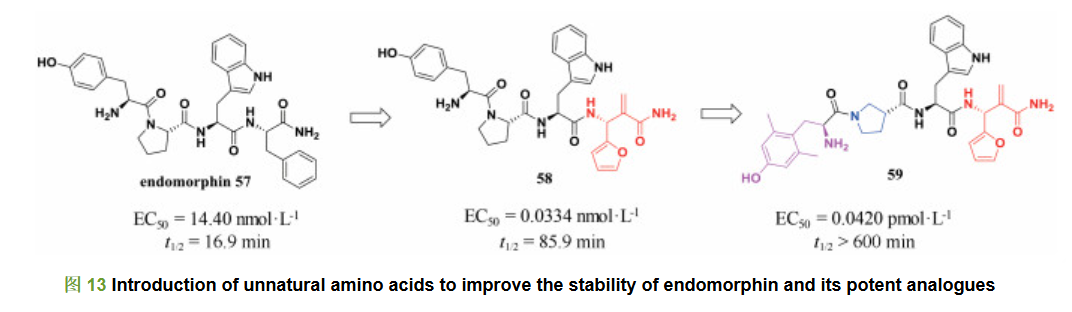
阿片受体与疼痛密切相关, 主要包括μ受体、δ受体和κ受体等几种亚型。阿片肽则是一种内源性神经递质, 通过与这些受体结合发挥药理作用。研究人员发现内吗啡肽57是μ受体的内源性底物肽, 具有较强的药理活性, 其对μ受体的激动活性为14.40 nmol・L-1, 相较于吗啡不会产生严重的不良反应; 而且, 内吗啡肽在有效剂量下不易诱发呼吸抑制和心血管疾病。因此, 内吗啡肽引起了科学家的广泛关注。然而, 内吗啡肽仍存在一些问题, 其中之一就是其代谢稳定性较差, 半衰期仅为16.9 min。兰州大学王锐团队发现含有非天然氨基酸的内吗啡肽类似物具有较强的代谢稳定性(图 13), 而且可以在一定程度上进一步提高内吗啡肽对μ受体的活性。他们首先将C末端苯丙氨酸替换为非天然氨基酸, 得到化合物58, 其对μ受体的激动活性为0.033 4 nmol・L-1, 相比于内吗啡肽57提高了430倍; 而且该化合物在脑膜匀浆中的半衰期延长至85.9 min, 与内吗啡肽相比提高了近4倍[29]; 随后, 他们在此工作的基础上进一步把酪氨酸和脯氨酸片段用非天然氨基酸替换, 得到化合物59, 其对μ受体的活性进一步提高, 达到0.042 0 pmol・L-1。而且化合物59在脑膜匀浆中的半衰期超过600 min[30], 解决了内源性吗啡肽半衰期短的问题。因此, 非天然氨基酸的引入对改善肽类化合物的代谢稳定性具有重要意义。
 天然多肽大多由L型氨基酸组成, 容易受到各种蛋白酶的降解而失去活性。蛋白酶的水解反应一般都是立体专一的, 引入D型氨基酸使多肽的构型发生变化, 进而使得修饰的多肽不易被蛋白水解酶水解, 因此D型氨基酸修饰的多肽可以提高对蛋白酶的降解作用。
天然多肽大多由L型氨基酸组成, 容易受到各种蛋白酶的降解而失去活性。蛋白酶的水解反应一般都是立体专一的, 引入D型氨基酸使多肽的构型发生变化, 进而使得修饰的多肽不易被蛋白水解酶水解, 因此D型氨基酸修饰的多肽可以提高对蛋白酶的降解作用。
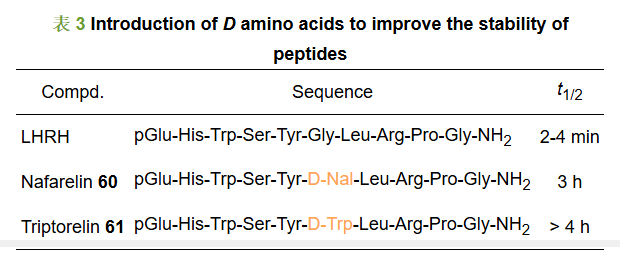
黄体激素释放激素(luteinizing hormone releasing hormone, LHRH)是由下丘脑分泌的具有调节生殖功能的十肽, 该激素与垂体前叶的黄体激素释放激素受体(gonadotropin-releasing hormone receptor, GnRHR)结合, 可以调控黄体激素的合成和分泌。除此之外, 在人类多种恶性肿瘤中, LHRH与其他生长因子一起调节肿瘤细胞生长。LHRH及类似物可以通过抑制垂体-性腺轴的功能从而抑制激素依赖性肿瘤细胞的增殖, 因此LHRH及类似物目前在临床上用于治疗激素依赖性肿瘤如前列腺癌和乳腺癌等。然而天然的LHRH第5、6位以及第6、7位氨基酸残基间肽键稳定性较差, 在体内极易受到肽链内切酶的作用而裂解, LHRH在体内的半衰期仅有2~4 min。为了提高LHRH在体内的稳定性, 研究人员尝试在6位引入不同种类的D型氨基酸, 得到上市药物如那法瑞林60和曲普瑞林61, 半衰期相较于LHRH均有不同程度的提高, 其半衰期分别为3 h和4 h (表 3)[31]。
2.1.2 伪肽化策略
肽键(-CONH2-)是肽类分子的特征, 而肽键在体内容易被蛋白酶识别降解, 这是肽类分子稳定性差的原因之一。伪肽则是利用生物电子等排原理将肽键中的一种或两种以上的原子用其他原子替代。由于伪肽从本质上改变了酰胺键的化学结构, 与蛋白或多肽同源结构不同, 因此可以避免体内蛋白酶的识别和水解, 从而提高肽类分子的稳定性及活性。
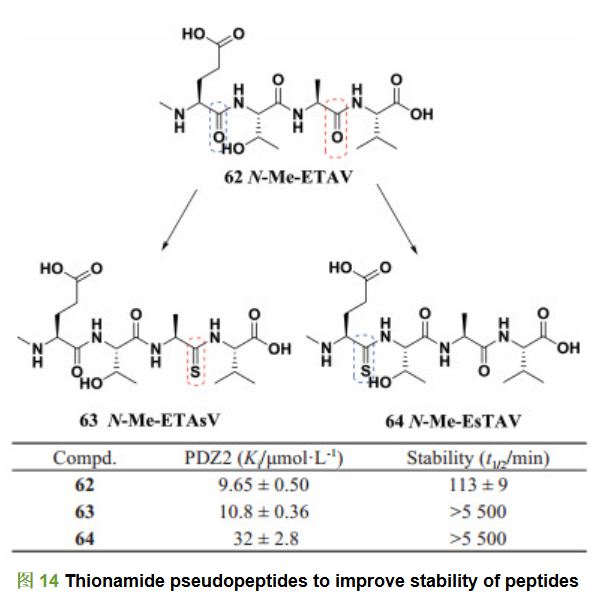
N-甲基-D-天冬氨酸(N-methyl-D-aspartate, NMDA)受体与其胞内突触后致密蛋白(postsynaptic density protein-95, PSD-95)的蛋白-蛋白相互作用是治疗缺血性脑病、神经疼痛以及阿尔兹海默症的一种潜在策略[32]。Bach等[33]报道, N-烷基化的谷氨酸-苏氨酸-丙氨酸-缬氨酸四肽化合物(N-甲基-ETAV) 62是NMDA/PSD-95蛋白-蛋白相互作用抑制剂(Ki=9.65 μmol・L-1), 他们通过结构修饰得到一系列活性较强的四肽衍生物, 但是研究人员在改造过程中发现这类化合物的血浆稳定性较差, 例如, 化合物62在人血浆中的半衰期只有113 min, 较差的代谢稳定性限制了该类化合物的进一步开发。为了改善化合物的血浆稳定性, Bach等对该类化合物进行伪肽化结构修饰, 将酰胺键的氧原子用硫原子进行替换, 得到不易被蛋白酶识别并水解的硫杂酰胺键。比较含硫杂酰胺键的伪肽63、64和含有酰胺键的化合物62, 可以发现含硫杂酰胺键的化合物虽然活性有所下降, 但血浆稳定性显著提高(图 14), 尤其是化合物63, 在活性基本不变(Ki=10.8 μmol・L-1)的同时血浆半衰期提高了50倍。研究结果表明, 硫杂酰胺键的伪肽化修饰是提高肽类化合物血浆稳定性的有效策略。
2.1.3 逆肽策略
蛋白质、激素、活性肽以及天然产物多肽是各种蛋白酶降解的底物, 因此存在着易受蛋白酶降解以及半衰期较短的特点。除了之前介绍的策略可以有效耐受蛋白酶的水解, 肽键方向的改变同样可以改变蛋白酶对底物的识别作用, 从而达到抗降解的作用。这类改变肽键方向的多肽结构修饰策略称为逆肽化修饰, 相关的肽称为逆肽或逆反肽。
β-淀粉样蛋白(amyloid β-protein, Aβ)沉积物的形成可能是引起AD的重要过程。研究表明Aβ可溶性寡聚体有细胞毒性, 并且对大脑的记忆能力和学习能力具有潜在影响。在Aβ聚集的早期进行抑制可有效治疗AD。Taylor等[34]报道了能有效抑制Aβ聚集的九肽65, 尽管65对Aβ寡聚体的聚集有较强的抑制作用, 但65存在多个水解位点, 因此需要对65进行结构修饰以提高其代谢稳定性。对65进行逆肽修饰, 得到逆肽66, 理论上逆肽可以保持与65相似的三维结构从而使活性得到保持, 实验结果也表明逆肽66对Aβ寡聚体的聚集抑制活性并没有发生显著变化。Taylor等用蛋白质降解实验评价65和66的代谢稳定性, 即将肽与人血浆或脑提取物共孵育24 h, 通过高效液相色谱法(HPLC)测定溶液中原型肽含量。可以发现无论血浆还是脑提取物中, 逆肽66的含量均远远高于65, 而且65的含量接近100%, 表明逆肽可以一定程度上提高化合物的代谢稳定性(图 15)。
 2.1.4 环化策略
2.1.4 环化策略
肽去甲酰基酶(peptide deformylase, PDF)是参与细菌蛋白质生物合成和成熟的重要酶, 在细菌和真核生物的细胞器中, 蛋白质的合成始于N-甲酰蛋氨酸, 因此新合成的多肽都含有甲酰化的N末端。PDF催化这些多肽的去甲酰化过程。PDF在细菌细胞中发挥的重要作用使其成为设计新型抗生素, 治疗耐药性病原体的新靶标。研究人员在前期工作基础上发现化合物67具有一定的抗菌活性, 其对大肠杆菌PDF抑制活性Ki值为92 nmol・L-1。但67在大鼠血浆中容易受到类胰蛋白酶的降解作用而失活。从图 16中可以发现, 67在大鼠血浆中孵育5 h约25%被降解。为了提高血浆稳定性, 研究人员将P1'与P3'进行环化, 设计合成环肽类似物68。研究结果表明, 相比于67, 环肽类似物68的抗菌活性有所提高, Ki值为74 nmol・L-1, 而且血浆稳定性大幅提高, 将68与大鼠血浆孵育5 h基本不被降解[35]。

α螺旋是大部分多肽分子都具有的二级结构特征, 然而人工合成的多肽分子在水溶液中并不能保持稳定的α螺旋结构[36], 因此科研人员开发了一种以碳-碳键或其他连接链为支撑的骨架稳定多肽α螺旋结构, 由这类方法得到的多肽称为订书肽(stapled peptide), 该方法本质上也属于环化修饰策略的一种。线性肽柔性大, 在舒展的构象下, 容易暴露出更多酶解位点, 增加了多肽被水解的概率, 从而导致多肽稳定性降低[37]。形成订书肽可以约束线性多肽的构象, 减少多肽被降解的概率。
β连环蛋白-B细胞淋巴瘤9 (B-cell lymphoma, BCL9)蛋白-蛋白相互作用对β连环蛋白的转录活性至关重要, 而这一相互作用是由BCL9蛋白中25个残基的螺旋片段和β连环蛋白的结合槽介导。王少萌等发现, 372位突变的BCL9肽69具有一定抑制β连环蛋白的活性(Ki=0.94 μmol・L-1), 然而69稳定性较差, 在细胞培养液中1 h降解75% (图 17)。因此王少萌等设计了一类结构稳定, 不容易被代谢的BCL9肽[38]。在设计过程中, 他们采用了点击化学(click chemistry)形成的三氮唑为支撑结构, 合成订书肽70和71。订书肽70和71对β连环蛋白的抑制活性分别为0.61 μmol・L-1和0.19 μmol・L-1, 活性保持。同时提高了线性肽的稳定性, 70和71在细胞培养液中1 h仅分别降解30%和25%。
 2.2 外接基团修饰
2.2 外接基团修饰
外接基团修饰以增强多肽分子代谢稳定性的主要方法包括高级脂肪酸修饰、蛋白融合策略、聚乙二醇修饰等。
2.2.1 高级脂肪酸修饰
高级脂肪酸修饰是指在肽类药物的特定位点通过化学方法以共价键的形式引入高级脂肪酸以改善肽类药物的性质, 延长半衰期。一般认为, 高级脂肪酸修饰可以稳定其结构, 提高多肽的稳定性, 从而延长多肽药物在体内的半衰期。同时, 高级脂肪酸与细胞膜表面的磷脂结构类似。因此, 脂肪酸修饰的多肽药物往往也可以提高多肽药物的脂溶性, 改善药物在肠道内的吸收以及黏膜透过性。此外, 高级脂肪酸可以与血清白蛋白(human serum albumin, HSA)结合, 结合后的复合体因分子过大而不容易转运, 从而可以延长多肽在体内的循环时间[39]。目前, 高级脂肪酸作为修饰结构的研究发展仍然比较缓慢, 但高级脂肪酸作为体内的一种内源性物质, 一直吸引了研究人员的广泛关注。根据水蛭素结构简化得到的水蛭肽比伐卢定72 (Bivalirudin)是由The Medicines Company开发的抗凝药物, 于2000年12月年被FDA批准上市, 作为抗凝剂用于经皮冠状动脉腔内成形术(percutaneous transluminal coronary angioplasty, PTCA)治疗中出现的不稳定型心绞痛和经皮冠状动脉介入治疗(percutaneous coronary intervention, PCI)。但是作为多肽类药物, 72在体内的暴露量较低(AUC0-t为23.7 nmol・min・mL-1), 半衰期短(t1/2 =15.1 min), 药代动力学性质较差。在进行经皮冠状动脉介入治疗之前, 需要先进行静脉注射, 随后静脉滴注至手术结束, 患者依从性差。针对这一缺点, 研究人员对比伐卢定类似物73进行化学修饰, 主要的策略是用高级脂肪酸对氨基酸侧链进行修饰。对比肽73和74, 其药理活性基本保持不变, 而高级脂肪酸修饰的多肽74暴露量(AUC0-t为1371.7 nmol・min・mL-1)和半衰期(t1/2=212.2 min)相较于未修饰的多肽73 (AUC0-t为25.7 nmol・min・mL-1, t1/2=13.5 min)明显改善(表 4), 暴露量和半衰期分别提高了58倍和14倍[40]。

上市的降糖多肽药物利拉鲁肽[41]和索马鲁肽[42]也都引入了高级脂肪酸修饰, 高级脂肪酸的引入增加了药物的疏水性, 掩盖二肽基肽酶4 (DPP-4)的结合位点, 降低肾排泄, 提高半衰期。利拉鲁肽是由诺和诺德公司研发的长效GLP-1受体激动剂, 其与天然GLP-1有97%的氨基酸序列相似性, 仅在34位将赖氨酸替换为精氨酸, 同时在26位赖氨酸侧链引入由谷氨酸作为linker的16碳棕榈酸侧链。皮下注射利拉鲁肽后, 其可在注射部位形成稳定的七聚体, 在皮下组织缓慢吸收; 另外, 由于引入了长链脂肪酸修饰, 掩盖了DPP-4结合位点; 同时, 长链脂肪酸的引入还使利拉鲁肽与血清白蛋白形成可逆复合物, 极大地延长了利拉鲁肽在体内的吸收时间, 提高了多肽类药物的体内半衰期。天然的GLP-1半衰期极短, 只有2 min左右; 而棕榈酸修饰的利拉鲁肽半衰期延长至13 h, 提高了390倍(图 18)[43, 44]。索马鲁肽则是GLP-1(7-37)的第8位丙氨酸用氨基异丁酸替换, 34位的赖氨酸用精氨酸替换, 同时在26位赖氨酸侧链由谷氨酸作为linker引入十八烷酸, 疏水性也更强, 同时经过短链的聚乙二醇修饰, 其半衰期大大延长至一周。
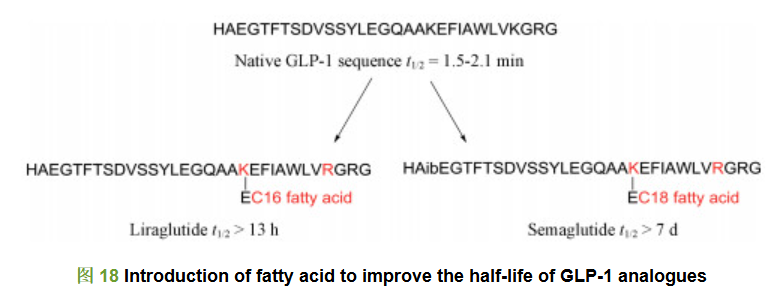
除了利用可逆的结合方式结合HSA, 共价不可逆的结合方式也常常用于多肽类药物的改造中。艾博卫泰(albuvirtide)是由南京前沿生物技术有限公司开发的全球首个长效抗HIV-1药物, 其结构如图 19所示。恩夫韦肽(enfuvirtide)是FDA批准的第一个临床使用的HIV-1融合抑制剂。然而恩夫韦肽75作为一个多肽药物, 其在人体内的半衰期只有3.5~4.4 h, 需要每天注射两次, 患者的依从性较差。针对恩夫韦肽75半衰期较短的缺点, 前沿生物技术有限公司开发了艾博卫泰, 艾博卫泰76是在多肽序列13位的赖氨酸侧链中引入了3-马来酰亚胺-丙酸(MPA)修饰(图 19), MPA可与血清白蛋白中的巯基形成不可逆的共价结合, 而且结合速率快, 大大提高了多肽在人体内的半衰期, 给药频率一周一次即可[45]。艾博卫泰已于2018年获得国家食品药品监督管理总局(CFDA)批准上市, 用于与其他抗逆转录病毒药物联合使用, 治疗HIV-1感染。

2.2.2 蛋白融合策略
蛋白融合策略是指利用基因工程技术, 将蛋白或多肽分子与免疫球蛋白Fc片段或血清白蛋白HSA融合而产生新型分子的修饰策略。融合Fc或HSA片段之后的多肽分子, 分子尺寸显著增大, 降低了肾对多肽药物的清除率, 从而延长多肽药物的半衰期[46]。礼来公司开发的降糖药物度拉糖肽(dulaglutide)就是将GLP-1与IgG4 (Fc)融合而成的长效降糖药物[47], 其生物半衰期大于90 h, 并且疗效不弱于利拉鲁肽, 其在2019年前三季度的销售额达到29.20亿美元, 超过利拉鲁肽(2019年前三季度销售额为24.47亿美元)。
人血清白蛋白是血浆中含量最丰富的蛋白质, 其半衰期长达19天, 因此HSA蛋白融合可以延长多肽药物的半衰期。葛兰素史克公司研发的长效降糖药物阿必鲁肽(albighztide)是第一个被FDA批准上市的HSA蛋白融合药物, 阿必鲁肽的半衰期长达6~10天[48]。因此, 蛋白融合策略是多肽药物长效化的有效手段。
2.2.3 聚乙二醇修饰
聚乙二醇(polyethylene glycol, PEG)在体内具有可降解、低毒性、无抗原性等特点, 是一种常见的肽类分子修饰方法。PEG修饰可以改善肽类分子的稳定性、减少蛋白酶的降解、不易被肾小球滤过, 从而提高多肽药物的稳定性, 延长药物的半衰期。目前已有诸多PEG修饰的多肽药物上市, 其中PEG修饰的干扰素α是这一结构修饰策略的成功案例。干扰素α可以有效地抑制或清除乙型肝炎或丙型肝炎病毒, 但干扰素α作为多肽类药物具有自身不可克服的缺点, 其半衰期短, 仅为4 h, 需要每天注射一次。为了克服这一缺点, 先灵葆雅研究所(Schering-Plough Research Institute, SPRI)致力于长效干扰素α的研究。他们分析了干扰素α半衰期较短的原因, 认为其分子过小容易被肾脏清除, 因此研究人员将PEG引入到干扰素α中(图 20), 而修饰后的干扰素α整体分子尺寸变大, 不易被肾小球滤过, 从而PEG修饰的干扰素α半衰期得到延长, 达到40 h[49]。另一方面, 由于PEG的引入掩盖了干扰素α与受体的结合, 降低了干扰素α的抗病毒活性, 因而先灵葆雅研究所对PEG的尺寸进行考察, 最终确定PEG的大小为12 kDa可以在延长半衰期的同时最大程度保留了干扰素α的抗病毒活性。因此, 采用PEG修饰策略要注意平衡半衰期和活性的关系。

免责声明:本文为行业交流学习,版权归原作者及原杂志所有,如有侵权,可联系删除。文章标注有作者及文章出处,如需阅读原文及参考文献,可阅读原杂志。

电话:0551-65177703 邮箱:pb@peptidesbank.com 地址:安徽省合肥市四川路868号云谷创新园A6栋3层
合肥肽库生物(Taikubio)只为有资质的科研机构、医药企业基于科学研究或药证申报的用途提供医药研发服务, 不为任何个人或者非科研性质的、非用于药证申报使用等其他用途提供服务。