多肽是由各种氨基酸分子之间脱水形成肽键相连的有机物, 其分子质量在1~10 kDa, 介于小分子和生物大分子之间。存在于体内的诸多信号分子都属于肽或蛋白质, 疾病的发生发展离不开这些肽或蛋白质。就目前已知的活性肽而言, 大部分都是由机体分泌或代谢转化而来。因此, 按照活性肽的来源可以将肽分为两类:第一类是来源于生物体本身的蛋白质及活性肽, 称为内源性活性肽。内源性活性肽在体内含量少、分布广、效应极强。第二类是来源于动植物的活性多肽以及抗生素等, 称为外源性活性肽。外源性活性肽作用强、分布广泛。内源性活性肽和外源性活性肽构成的多肽库为药物研发提供了新颖的结构骨架, 许多上市药物都源自多肽化合物的发现。
是经典的多肽结构对体内蛋白酶的稳定性较差, 进入体内很快会被降解; 此外, 大多生物活性肽生物利用度比较差, 无法口服, 需要通过改变剂型进而研发获取适合的给药途径。基于以上这些因素, 需要对活性肽进行结构修饰与化学改造[1]。活性肽改造的目的多种多样, 主要包括提高活性肽与受体的亲和力及选择性; 增强多肽分子的药代稳定性, 降低活性肽在体内的降解或者减少活性肽在体内的消除; 提高活性肽的透膜能力; 改善疏水肽的水溶性等。本文针对不同改造目的总结归纳了肽类分子结构修饰改造策略, 根据是否对肽链骨架进行修饰, 将这些修饰策略分为两类:一类是针对肽链骨架的改造, 包括非天然氨基酸修饰、伪肽化策略、逆肽策略、环化策略、末端结构修饰等; 另一类是在多肽骨架不变的基础上, 引入其他基团进行结构优化和性能改造, 包括高级脂肪酸修饰、聚乙二醇修饰、蛋白融合策略、胆固醇修饰等。通过综合运用这些先导化合物结构修饰策略, 能够显著提高多肽类化合物的成药性, 为开发多肽类创新药物提供理论指导和实践经验。
1 提高肽类分子活性
药物的化学结构与药理活性之间的关系一直都是药物化学领域的重要研究内容。活性是化合物开发成药物的前提, 多肽也是如此。部分天然肽类分子或人工合成的肽生物活性差, 需要通过化学修饰提高肽类分子与受体的亲和力, 改善肽的活性。提高肽类分子活性的主要方法包括末端结构修饰、拼接策略、环化策略、非天然氨基酸修饰、伪肽策略以及胆固醇修饰等。
1.1 肽链骨架改造
对肽链骨架进行修饰和改造以提高肽类分子活性的主要方法包括末端结构修饰、拼接策略、环化策略、非天然氨基酸修饰、伪肽策略等。
1.1.1 N-Cap结构修饰
N端裸露和C端裸露的多肽容易受到肽链外切酶的识别, 从而被切割降解失去活性。而将N末端和C末端进行结构修饰, 一方面可以提高肽类分子的代谢稳定性, 另一方面可以保持甚至提高肽类分子的活性。Stoermer等[2]报道了三肽(KKR序列)醛类化合物作为西尼罗病毒(west Nile virus, WNV)蛋白酶抑制剂; 尹正课题组报道了三肽(KRR序列)醛类化合物作为登革热病毒(Dengue virus, DENV)蛋白酶抑制剂, 并且相较于其他四肽醛类化合物活性显著提高[3]。基于此类研究报道, Andreas等[4]认为不同氨基末端结构修饰的三肽醛类化合物对活性有不同影响, 因此他们对N端Cap区进行考察, 得到不同酰基化修饰的三肽醛类化合物, 并且发现不同酰基化修饰对化合物的抗病毒活性有较大影响(表 1)[4], 可以看出N端苯乙酰基修饰的三肽醛2对登革热病毒和西尼罗病毒都有较好的抑制活性, 而N端4-苯基苯乙酰基修饰的三肽醛11相比于2对西尼罗病毒的抑制活性提高近7倍。这一点表明氨基末端的结构修饰对肽类化合物的活性有一定影响, 可以作为肽类化合物改造的一种策略。

1.1.2 C-末端结构修饰
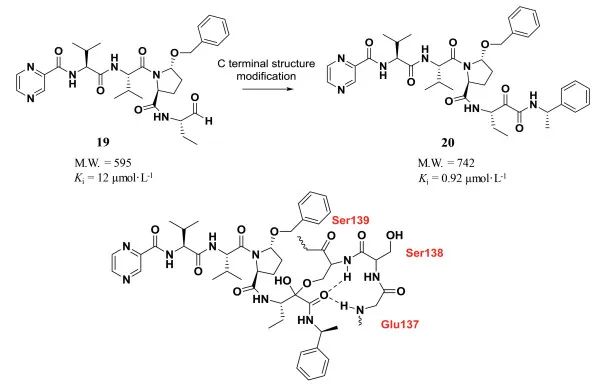
与氨基端结构修饰相对应, 羧基端结构修饰在肽类分子的修饰改造也具有广泛应用, C-末端结构修饰策略成功地在各类病毒蛋白酶抑制剂的结构改造中使用。丙肝病毒NS3/4A蛋白酶是一种丝氨酸蛋白酶, 目前大部分丝氨酸蛋白酶抑制剂都含有亲电基团, 与催化三联体的丝氨酸羟基形成共价键。研究人员从十肽底物出发经过肽链的简化以及羧基末端结构修饰得到活性肽醛19, 其对HCV NS3/4A蛋白酶的结合常数为12 μmol・L-1, 但醛基的化学和代谢稳定性较差。因此, 研究人员对醛基末端进行结构修饰, 以α-卤代酮、杂代酮、α-二酮和α-酮酰胺替换醛基, 得到的酮酰胺化合物20对丝氨酸蛋白酶NS3/4A的结合常数提高了12倍。分析酮酰胺20与丝氨酸蛋白酶的相互作用(图 1), 研究人员发现酮酰胺结构既可以与139位丝氨酸形成共价结合, 又可以与附近的137位谷氨酸和138位丝氨酸残基形成氢键作用, 增强了化合物与NS3/4A蛋白酶的结合, 因而其抗病毒活性提高[5]。
EV71 3C蛋白酶是一种半胱氨酸蛋白酶。尹正等[6]发现了对EV71病毒具有较好抑制活性的肽醛分子21 (EC50=0.11 μmol・L-1), 考虑到醛基的稳定性较差, 成药性质不佳。他们在进一步的结构修饰中, 针对醛基进行结构优化, 得到羟基氰类化合物22, 该化合物对EV71的活性为0.056 μmol・L-1。通过分子对接, 分析化合物22与EV71 3C蛋白酶的相互作用, 分子对接结果表明(图 2), 相比于醛基, 羟基氰结构中的氰基与146位谷氨酰胺和24位谷氨酸通过水分子形成氢键, 增强了化合物与EV71 3C蛋白酶的结合, 因而活性得以提高[6, 7]。

1.1.3 拼接策略
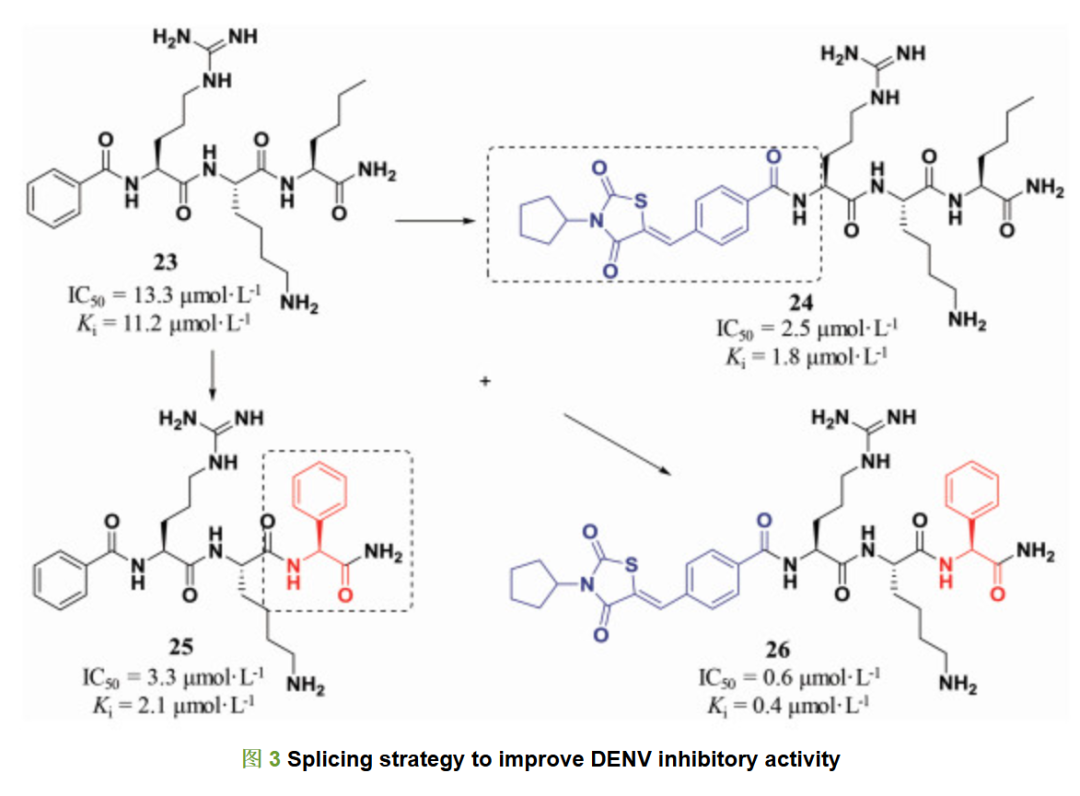
在肽类化合物的改造中, 往往需要对不同位点同时进行优化和改造, 拼接策略是一个高效的结构优化方法。首先, 分别对N端和C端结构修饰改造得到活性较优的化合物, 然后将优势片段进行拼接, 即可快速获得活性更高的化合物。拟肽化合物23是一个登革热病毒蛋白酶抑制剂, 其抑制活性IC50为13.3 μmol・L-1。在对该化合物改造的过程中, 研究人员就采用了分别优化N端和C端的研究策略。在N端结构改造中, 研究人员发现肽类分子24, 即N端Cap结构修饰的化合物, 活性提升, 其IC50达到2.5 μmol・L-1。在C端侧链改造过程中研究人员也发现将正丁基侧链替换为苯基得到化合物25, 同样可以提高化合物对登革热病毒的抑制活性, 活性提升近4倍。考虑到这两个修饰策略都可以提高化合物的活性, 研究人员将两个优势片段组合拼接, 得到化合物26, 其对登革热病毒的抑制活性为0.6 μmol・L-1, 活性提高了近20倍(图 3)[8]。
 1.1.4 环化策略
1.1.4 环化策略
许多情况下, 直链肽的分子柔性造成构象发生变化, 使其与受体结合的强度及选择性下降。此外, 生物体内的氨肽酶及羧肽酶也易于从直链肽两个端基逐步切割肽链, 使之降解。因此肽链的环化改造, 使其构象限定是改善肽类分子生物稳定性、提高生物活性的重要结构改造策略[9]。研究表明, 从直链肽改为环肽后, 许多化合物的生物活性提高十几倍至几万倍。许多具有抗菌、抗病毒、抗肿瘤、免疫调节等活性的天然产物肽往往含有不同类型的主链环化结构。因此, 环化策略是多肽结构修饰改造的一个重要策略。
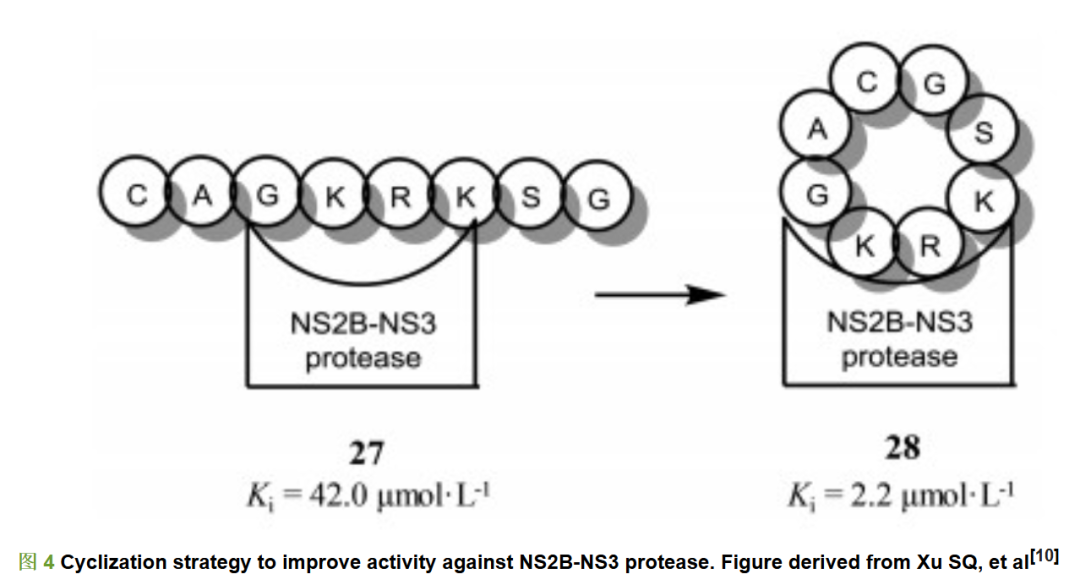
线性八肽化合物27对登革热病毒NS2B-NS3蛋白酶有较弱的结合活性(Ki=42 μmol・L-1)。Xu等[10]推测线性肽结合活性较差的原因可能是线性肽占用的空间较大, 无法与蛋白酶有效地结合; 而用环肽则可以改变线性肽所占用的空间, 提高化合物对NS2B-NS3蛋白酶的结合活性。他们设计合成了系列环肽结构, 并且对这类环肽的结合活性进行测试。发现环肽28的构象使得其可以较好地与登革热病毒NS2B-NS3蛋白酶结合, 相较于线性肽活性提高近20倍, Ki值达到2.2 μmol・L-1 (图 4)。
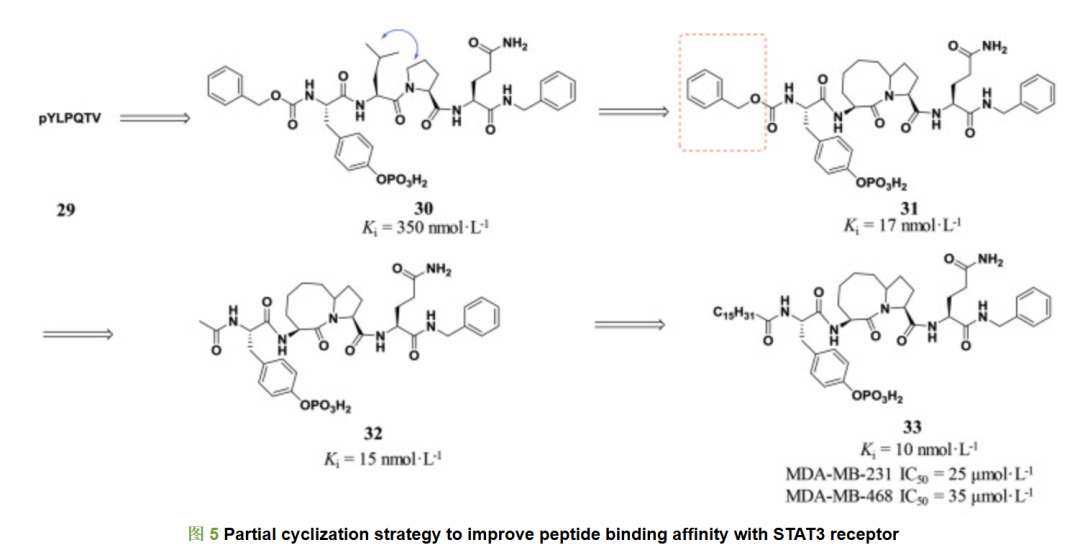
 除了首尾相连的大环化策略, 局部环化往往能够局部限定环化区域的肽类化合物构象, 稳定肽类化合物与受体的相互作用, 提高肽类化合物的活性。信号传导及转录激活因子STAT3是一种直接将细胞外受体的信号传递至核内的转录因子。STAT3的持续激活会促进细胞增殖从而形成肿瘤并且抑制肿瘤细胞凋亡[11-13]。美国密歇根大学王少萌等[14]早期发现gp130 pYLPQTV肽29与STAT3受体有较强的亲和力。研究表明, gp130磷酸肽中的苏氨酸和缬氨酸可以被苄胺替代而不改变肽和STAT3之间的结合活性, 因此, 研究人员切断苏氨酸和缬氨酸并用苄胺封闭碳端, 得到了截断磷酸肽30, 30与STAT3受体的结合力Ki为350 nmol・L-1。通过分子对接, 他们发现亮氨酸侧链异丁基和脯氨酸五元环可以并环形成双环内酰胺结构而不破坏肽30 β转角的构象。因此, 采用局部环化策略, 他们设计合成了构象限制的双环类肽化合物31, 其与STAT3受体的结合力Ki为17 nmol・L-1, 活性提高了近20倍。分子对接结果表明, 双环内酰胺结构可以很好保持30的β转角构象。为了验证Cbz保护基是否和肽31与STAT3受体结合相关, 他们将苄氧羰基替换为乙酰基, 得到的肽32与STAT3受体的结合力Ki值为15 nmol・L-1, 与肽31活性相当, 说明Cbz并非活性必须。随后, 他们评价了肽32对两种含有高磷酸化STAT3受体的人乳腺癌细胞株MDA-MB-231和MDA-MB-468的抑制活性, 但32在100 μmol・L-1水平下对这两种肿瘤细胞并没有表现出抑制活性, 可能是磷酸肽的极性太大, 无法通过细胞膜。为了增强32对肿瘤细胞的抑制活性, 他们将高级脂肪酸引入磷酸肽的氮端, 得到了脂肪酸修饰的磷酸肽33, 33与STAT3受体的结合力Ki值为10 nmol・L-1, 而且肽33对两种细胞的抑制活性IC50分别为25和35 μmol・L-1, 在细胞水平显示了一定的抑制活性, 也表明脂肪酸修饰可以改变肽类化合物的性质, 提高肽类化合物的透膜性(图 5)[15]。
除了首尾相连的大环化策略, 局部环化往往能够局部限定环化区域的肽类化合物构象, 稳定肽类化合物与受体的相互作用, 提高肽类化合物的活性。信号传导及转录激活因子STAT3是一种直接将细胞外受体的信号传递至核内的转录因子。STAT3的持续激活会促进细胞增殖从而形成肿瘤并且抑制肿瘤细胞凋亡[11-13]。美国密歇根大学王少萌等[14]早期发现gp130 pYLPQTV肽29与STAT3受体有较强的亲和力。研究表明, gp130磷酸肽中的苏氨酸和缬氨酸可以被苄胺替代而不改变肽和STAT3之间的结合活性, 因此, 研究人员切断苏氨酸和缬氨酸并用苄胺封闭碳端, 得到了截断磷酸肽30, 30与STAT3受体的结合力Ki为350 nmol・L-1。通过分子对接, 他们发现亮氨酸侧链异丁基和脯氨酸五元环可以并环形成双环内酰胺结构而不破坏肽30 β转角的构象。因此, 采用局部环化策略, 他们设计合成了构象限制的双环类肽化合物31, 其与STAT3受体的结合力Ki为17 nmol・L-1, 活性提高了近20倍。分子对接结果表明, 双环内酰胺结构可以很好保持30的β转角构象。为了验证Cbz保护基是否和肽31与STAT3受体结合相关, 他们将苄氧羰基替换为乙酰基, 得到的肽32与STAT3受体的结合力Ki值为15 nmol・L-1, 与肽31活性相当, 说明Cbz并非活性必须。随后, 他们评价了肽32对两种含有高磷酸化STAT3受体的人乳腺癌细胞株MDA-MB-231和MDA-MB-468的抑制活性, 但32在100 μmol・L-1水平下对这两种肿瘤细胞并没有表现出抑制活性, 可能是磷酸肽的极性太大, 无法通过细胞膜。为了增强32对肿瘤细胞的抑制活性, 他们将高级脂肪酸引入磷酸肽的氮端, 得到了脂肪酸修饰的磷酸肽33, 33与STAT3受体的结合力Ki值为10 nmol・L-1, 而且肽33对两种细胞的抑制活性IC50分别为25和35 μmol・L-1, 在细胞水平显示了一定的抑制活性, 也表明脂肪酸修饰可以改变肽类化合物的性质, 提高肽类化合物的透膜性(图 5)[15]。
1.1.5 非天然氨基酸修饰
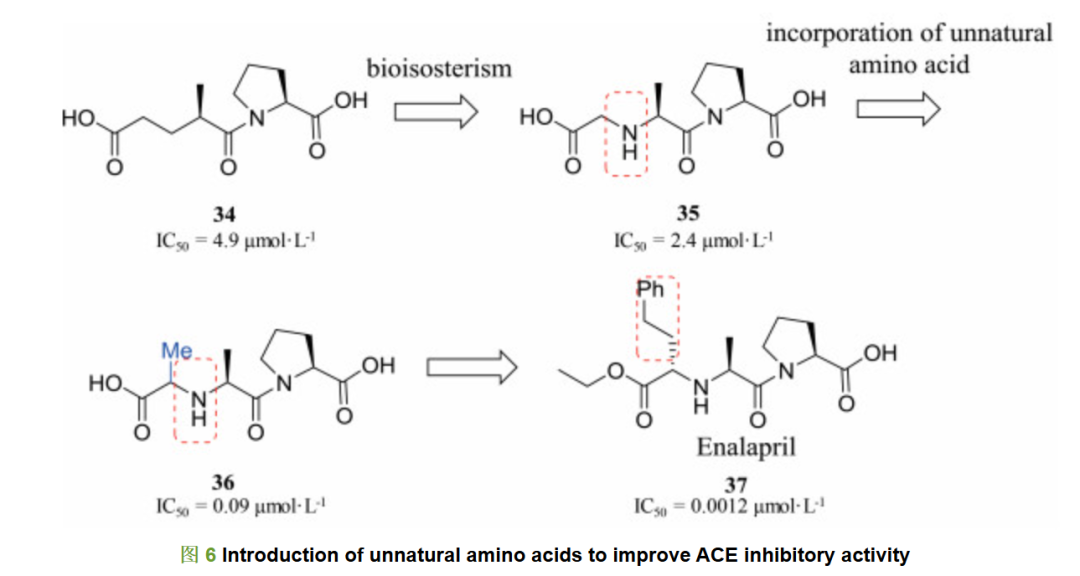
卡托普利是第一个报道的血管紧张素转化酶(angiotensin converting enzyme, ACE)抑制剂, 1981年被美国食品药品管理局(FDA)批准用于治疗高血压。临床研究表明, 卡托普利的巯基可能会引起患者皮疹和食欲减退等不良反应。为了解决这一问题, 研究人员研发新型ACE抑制剂作为降压药物。在卡托普利研发早期, 活性化合物34具有一定的ACE抑制活性, 其IC50为4.9 μmol・L-1。对34的亚甲基用氮原子进行生物电子等排, 得到二肽先导化合物35, 其活性提高1倍[16]。由于氮原子的引入, 化合物的亲水性有所增强; 为了平衡氮原子引起的亲水性增强, 研究人员尝试在氨基酸的α位引入烷基侧链平衡亲水性变化, 结果得到的化合物36活性进一步增强至0.09 μmol・L-1。随后, 研究人员对α位烷基侧链进行了详细的构效关系考察, 最终确定苯乙基取代时, 活性最优[17], 将羧基乙酯化开发获得前药依那普利37, 依那普利于1985年被美国FDA批准用于高血压和心力衰竭的治疗。37对ACE的抑制活性相比于36活性提高了74倍, 表明非天然氨基酸的引入可以增强肽类分子的药理活性(图 6)。
1.1.6 伪肽策略
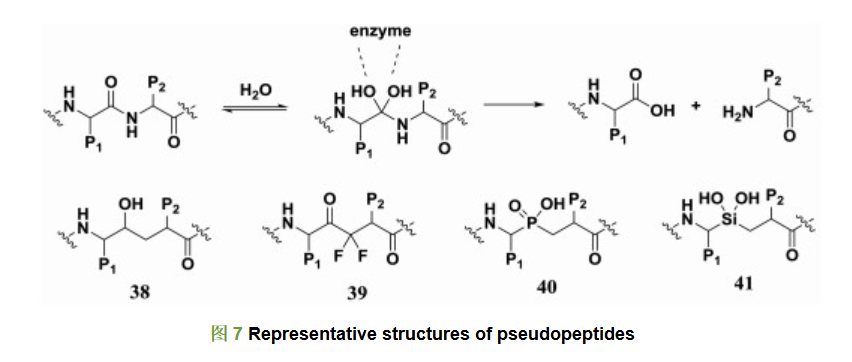
伪肽则是通过模拟多肽水解的过渡态, 利用生物电子等排原理对易水解的酰胺键进行替换, 使多肽免于蛋白酶的水解切割从而保留甚至提高肽类化合物的药理活性。图 7列出了一些伪肽的代表结构。
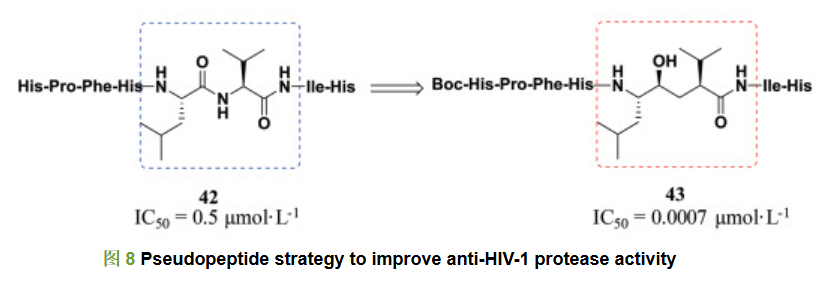
片段38 (羟基亚甲基)是众多HIV蛋白酶抑制剂、肾素抑制剂和β-分泌酶抑制剂[19]共有的结构片段。其基本的设计原理就是利用伪肽策略, 模拟酰胺键水解过程中的过渡态, 替换易水解的酰胺键。Szelke等[20]通过在肾素底物42的亮氨酸-缬氨酸(Leu-Val)片段中采用羟基亚甲基替换酰胺键, 得到伪肽抑制剂43, 对HIV-1蛋白酶抑制活性显著提高(图 8), 其IC50值为0.000 7 μmol・L-1。
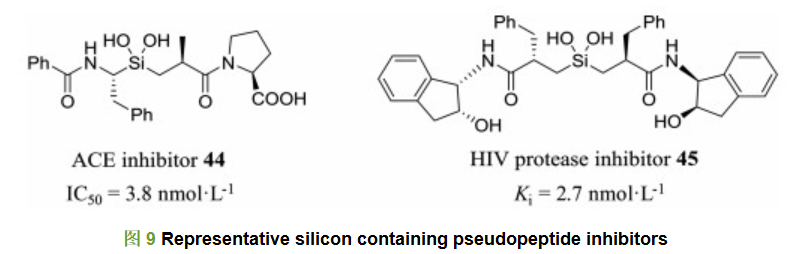
 其中缩硅酮片段41也有广泛应用, 由于碳原子和硅原子同属一个主族, 两个原子的性质十分相似, 而硅原子相比碳原子更倾向于sp3杂化, 片段41不容易发生消除反应生成硅酮, 缩硅酮的稳定性要高于缩酮, 因此在设计和改造活性肽的时候引入41片段既可以增强与酶活性中心的相互作用, 又具有一定的化学稳定性, 在药物化学化合物设计中具有广泛应用(图 9)。片段41在很多活性肽类似物分子上显示出良好活性, 例如含有片段41的ACE抑制剂44, 其对ACE酶的抑制活性达到了3.8 nmol・L-1; 而含有片段41的HIV蛋白酶抑制剂45对蛋白酶的抑制活性也达到2.7 nmol・L-1, 表明该类结构在活性肽结构改造中有重要意义。
其中缩硅酮片段41也有广泛应用, 由于碳原子和硅原子同属一个主族, 两个原子的性质十分相似, 而硅原子相比碳原子更倾向于sp3杂化, 片段41不容易发生消除反应生成硅酮, 缩硅酮的稳定性要高于缩酮, 因此在设计和改造活性肽的时候引入41片段既可以增强与酶活性中心的相互作用, 又具有一定的化学稳定性, 在药物化学化合物设计中具有广泛应用(图 9)。片段41在很多活性肽类似物分子上显示出良好活性, 例如含有片段41的ACE抑制剂44, 其对ACE酶的抑制活性达到了3.8 nmol・L-1; 而含有片段41的HIV蛋白酶抑制剂45对蛋白酶的抑制活性也达到2.7 nmol・L-1, 表明该类结构在活性肽结构改造中有重要意义。
1.2 外接基团修饰
外接基团修饰以提高肽类分子活性的主要方法是胆固醇修饰。
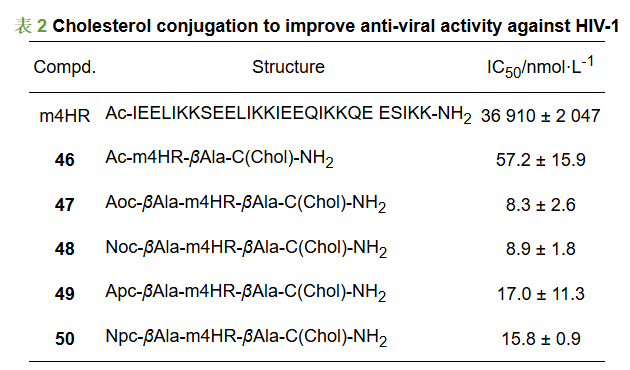
胆固醇修饰也是多肽类分子的重要结构修饰策略。胆固醇的引入常常可以在提高其在体内半衰期的同时增强多肽的药理活性。Wang等[22]用细胞-细胞融合实验评价多肽分子的抗病毒活性, 发现多肽m4HR具有一定抗HIV-1活性(IC50 =36 910 nmol・L-1)。当在m4HR C末端外接胆固醇分子得到化合物46, 其抗病毒活性提高200倍(IC50 =57.2 nmol・L-1)。进一步在N端修饰, 得到的肽类化合物对HIV-1的抑制活性进一步提升(表 2)。其中活性最好的是肽类分子47, 其IC50达到8.3 nmol・L-1, 该类化合物结构优化的实例进一步证明了胆固醇修饰在多肽药物活性优化的重要应用。

电话:0551-65177703 邮箱:pb@peptidesbank.com 地址:安徽省合肥市四川路868号云谷创新园A6栋3层
合肥肽库生物(Taikubio)只为有资质的科研机构、医药企业基于科学研究或药证申报的用途提供医药研发服务, 不为任何个人或者非科研性质的、非用于药证申报使用等其他用途提供服务。