
’Σ“Σ ΚΘ―σ «ΒΊ«ρ…œΉ ‘¥ΉνΈΣΖαΗΜΒΡΝλ”ρ. ΚΘ―σ…ζΈο…ζ¥φΜΖΨ≥Η¥‘”, ΙΥϋΟ«–Έ≥…ΝΥΨΏ”–ΧΊ βΫαΙΙΒΡΜν–‘Έο÷ ; Φ”÷°ΩΤΦΦΒΡΫχ≤Ϋ, »ΥΟ«“≤»’“φΙΊΉΔ»γΚΈ¥”ΚΘ―σ…ζΈο÷–―ΑΜώ–¬ΒΡΜν–‘Έο÷ , ’β ΙΒΟΚΘ―σ…ζΈοΜν–‘κΡ ήΒΫΙψΖΚΙΊΉΔ. …ζΈοΜν–‘κΡΨΏ”–Εύ÷÷Μν–‘, Ψ≠―–ΨΩ±μΟς, ΗΟάύΈο÷ ΨΏ”–ΩΙ―θΜ·ΓΔΩΙΗΏ―Σ―ΙΓΔΩΙ≤ΓΕΨΓΔΩΙ÷ΉΝωΒ»Μν–‘; «“ΨΏ”–ΒΆΕΨΓΔΗΏΕ»Α–œρΉ®“Μ–‘ΚΆ…ζΈοΜν–‘«ΩΒ»”≈Βψ. ΡΩ«Α, ¥”ΚΘΆΟΓΔ”σ¬ίΓΔΚΘ« ΓΔΚΘΟύΓΔΚΘ―σ’φΨζΓΔ»μΧεΕ·Έο÷–Χα»Γ≥ωΒΡ…ζΈοΜν–‘κΡΜράύΥΤΈο, ≤ΩΖ÷“―…œ –Μρ“―Ϋχ»κΝΌ¥≤ ‘―ιΫΉΕΈ. ’κΕ‘’β–©ΚΘ―σ…ζΈοΜν–‘κΡΒΡ―–ΨΩœ÷Ή¥, ΨΆΤδά¥‘¥ΓΔΚœ≥…ΖΫΖ®ΓΔΜ·―ßΫαΙΙΧΊΒψΓΔΜν–‘ΓΔΉς”ΟΜζ÷Τ“‘ΦΑΝΌ¥≤”––ß–‘ΦΑΑ≤»Ϊ–‘Β»ΖΫΟφΫχ––Ήέ ω, ≤Δ’ΙΆϊΝΥΗΟΝλ”ρΫώΚσΒΡΖΔ’ΙΖΫœρ.
ΚΘ―σΟφΜΐ‘Φ’Φ»Ϊ«ρΉήΟφΜΐΒΡ 70%, ’ΦΨί 90%ΒΡ…ζΈο»Π, ΚΘ―σ÷–…ζΈο÷÷άύ¥οΒΫ»Ϊ«ρ…ζΈοΕύ―υ–‘ΒΡΑκ ΐ, “ρ¥ΥΚΘ―σ «“ΜΗωΖαΗΜΒΡΧλ»ΜΜν–‘Μ·ΚœΈοΩβ[1]. ΚΘ―σΨΏ”–ΧΊ βΒΡΈοάμΚΆΜ·―ßΧθΦΰ[2], »γΗΏ―ΈΓΔ»θΦν, …νΚΘ«χ…θ÷ΝΨΏ”–ΚΎΑΒΓΔΚ°άδΓΔΗΏ―ΙΒ»Η¥‘”ΧΊΒψ; Φ”÷°Έο÷÷÷°Φδ«ΩΝ“ΒΡ…ζ¥φΨΚ’υ, ΙΒΟΚΘ―σ…ζΈο≥Θ≤…»ΓΜ·―ß ÷ΕΈΫχ––Ή‘Έ“±ΘΜΛ[3]; ÷πΫΞ–Έ≥…ΝΥ≤ΜΆ§”Ύ¬ΫΒΊ…ζΈοΒΡΜν–‘Έο÷ , »γΜν–‘κΡΓΔΕύ‘Σ≤Μ±ΞΚΆ÷§ΖΨΥαΓΔΙΧ¥ΦάύΓΔΟΗΓΔΕύΧ«ΓΔΩΙ―θΜ·ΦΝΚΆ…ΪΥΊΒ»[4]. ΥφΉ≈≈ύ―χΦΦ θΓΔΖ÷Ή”…ζΈο―ßΦΦ θΒΡΖΔ’Ι[5], ¥”ΚΘ―σ…ζΈο÷–Μώ»Γ–¬ΒΡΜν–‘Μ·ΚœΈο≤ΜΕœ»ΓΒΟ–¬Ϋχ’Ι, ”»Τδ «ΡΎ‘¥–‘ΕύκΡΜν–‘Ής”ΟΒΡΖΔœ÷“‘ΦΑΧλ»Μ…ζΈοΜν–‘κΡΒΡΑ–œρΉς”ΟΜζ÷ΤΒΡ―–ΨΩ, ΙΒΟκΡάύ≥…ΈΣΚΘ―σΜν–‘Έο÷ ΒΡ Ή“ΣΚρ―ΓΉ ‘¥[6].
…ζΈοΜν–‘κΡ”… 3ΓΪ20 ΗωΑ±ΜυΥα≤–ΜυΙΙ≥…, ΨΏ”–Εύ÷÷…ζΈοΜν–‘[7], »γΩΙ―θΜ·–‘ΓΔΩΙΗΏ―Σ―ΙΓΔΩΙ HIV ≤ΓΕΨΓΔΩΙ‘ω÷≥ΓΔΩΙΡΐ―ΣΓΔΗΤάκΉ”ρϋΚœΓΔΩΙΖ ≈÷ΓΔΩΙΧ«Ρρ≤ΓΒ»Μν–‘, ΤδΜν–‘”…Ζ÷Ή”¥σ–ΓΓΔΑ±ΜυΥα÷÷άύΚΆ¥Έ–ρΨωΕ®[8]. “ρΤδΨΏ”–ΒΆΕΨΓΔΗΏ–ßΓΔΗΏ―Γ‘ώ–‘Β»”≈ΒψΕχ±Μ”Π”Ο”Ύ ≥ΤΖ”κ÷Τ“©Νλ”ρ[9]. ΡΩ«Α, ΙζΡΎΆβΕ‘ΚΘ―σ…ζΈοΜν–‘κΡΫχ––ΝΥ¥σΝΩ―–ΨΩ. ΚΘ―σ…ζΈοΜν–‘κΡά¥‘¥÷ς“Σ”–ΚΘΟύΓΔΚΘ« ΓΔΚΘΩϊΓΔ”σ¬ίΓΔΚΘ‘εΓΔ”ψάύΓΔ»μΧεΕ·ΈοΓΔΦΉΩ«άύΕ·Έο, ΚΘ―σœΗΨζΚΆ’φΨζΒ»[6,10]. œ÷‘Ύ–μΕύ―–ΨΩ»Υ‘±¥”ΚΘ―σ…ζΈο÷–Χα»ΓΝΥ–μΕύΜν–‘ΫœΗΏΒΡΧλ»ΜΜν–‘κΡ, »γ Lee Β»[11]¥”¥σ¬μΙΰ”ψ÷–Ά®Ιΐ“»ΒΑΑΉΟΗΥ°ΫβΖ÷άκΒΟΒΫΨΏ”–“÷÷Τ―ΣΙήΫτ’≈ΥΊ I ΉΣΜ·ΟΗΒΡΜν–‘κΡ , ΤδΑ±ΜυΥα–ρΝ–ΈΣ Gly-Leu-Pro-Leu-AsnLeu-Pro, Κσ”÷“‘Χλ»ΜΒΡΜν–‘κΡΈΣΜυ¥ΓΚœ≥…ΝΥΜν–‘ΗϋΗΏΒΡ»ΐκΡ Gly-Leu-Pro, IC50÷ΒΈΣ 9.08 µmol•LΘ≠1; Zhan Β»[12]¥”ΚΘΟύ÷–Χα»Γ≥ωΈε÷÷–¬–ΆΜΖ–ΈκΡ reniochalistatins AΓΪE, Τδ÷– reniochalistatin E Ε‘ RPMI-8226 œΗΑϊΚΆ MGC-803œΗΑϊ”–œΗΑϊΕΨ–‘Ής”Ο, IC50÷ΒΖ÷±πΈΣ 4.9 ΚΆ 9.7 µmol•LΘ≠1. ”…¥ΥΩ…“‘Ω¥≥ω, “Μ–©ΚΘ―σ…ζΈοΜν–‘κΡ”–“ΜΕ®ΒΡ≥…“©«±ΝΠ. ”»Τδ «ΫϋΡξά¥, ΚΘ―σΧΫ≤βΦΦ θΒΡΖΔ’Ι, ΙΒΟΚΘ―σ“©ΈοΒΡΩΣΖΔΒΟΒΫΝΥΨό¥σΫχ’Ι. ΡΩ«Α, ¥”ΚΘΆΟΓΔ”σ¬ίΓΔΚΘ« ΓΔΚΘΟύΓΔΚΘ―σ’φΨζΓΔ»μΧεΕ·Έο÷–Χα»ΓΒΡΨΏ”–Μν–‘ΒΡΧλ»ΜκΡ, Μρ“‘Χλ»ΜκΡΈΣΡΗΧεΚœ≥…ΒΡ―ή…ζΈο“―Ϋχ»κ»Ϊ«ρΒΡ“©Έο –≥ΓΦΑΝΌ¥≤ ‘―ιΫΉΕΈ[13]. ±ΨΈΡΫΪΉέ ω“―…œ –Μρ¥Π”ΎΝΌ¥≤ ‘―ιΫΉΕΈΒΡΚΘ―σ…ζΈοΜν–‘κΡ, ≤ΔΑ¥’’Μν–‘Μ·ΚœΈοά¥‘¥Ζ÷άύ, Ϋι…ήΜν–‘Μ·ΚœΈοΜ·―ßΫαΙΙΧΊΒψΓΔΚœ≥…ΓΔΜν–‘ΓΔΉς”ΟΜζ÷Τ“‘ΦΑΡΆ ή–‘ΚΆΕΨΗ±Ής”ΟΒ», œΘΆϊΕ‘¥” ¬¥Υάύ―–ΨΩΒΡΩΤ―–»Υ‘±ΧαΙ©“ΜΕ®ΒΡ≤ΈΩΦ“άΨί.
1 ΚΘΆΟΕΨΥΊ
Pettit Β»[14]”Ύ 1972 ΡξΩΣ Φ’ΙΩΣΝΥΕ‘”ΓΕ»―σ÷–ΚΘΆΟΒΡΧα»ΓΈοΥυΚ§ΩΙ÷ΉΝωΜν–‘≥…Ζ÷ΒΡ―–ΨΩ, ΥϊΟ«ΖΔœ÷ΤδΧα»ΓΈοΩ…“‘÷ΈΝΤ–Γ σP388ΝήΑΆœΗΑϊΑΉ―Σ≤Γ, ―”≥Λ–Γ σ ΌΟϋ. ΥφΚσΥϊΟ«”÷Ζ÷άκΒΟΒΫ“ΜœΒΝ–ΨΏ”–“÷÷ΤœΗΑϊ‘ω÷≥ΓΔΩΙ÷ΉΝωΜν–‘ΒΡκΡάύΜ·ΚœΈο, ΟϋΟϊΈΣ Dolastatin, Τδ÷–Μν–‘Ήν«ΩΒΡ «Dolastatin 10 ΚΆ Dolastatin 15. ¥ΥΚσ, –μΕύ―–ΨΩ’ΏΕ‘Dolastatin 10 ΚΆ Dolastatin 15 ’ΙΩΣΝΥΫχ“Μ≤Ϋ―–ΨΩ, ≤ΔΚœ≥…ΝΥ“ΜœΒΝ–―ή…ζΈο. Τδ÷–, “‘ auristatin œΒΝ–ΓΔTasidotinΚΆ LU-103793 Μν–‘ΉνΚΟ, ”Ύ 1995 ΡξΫχ»κΝΌ¥≤ ‘―ι.
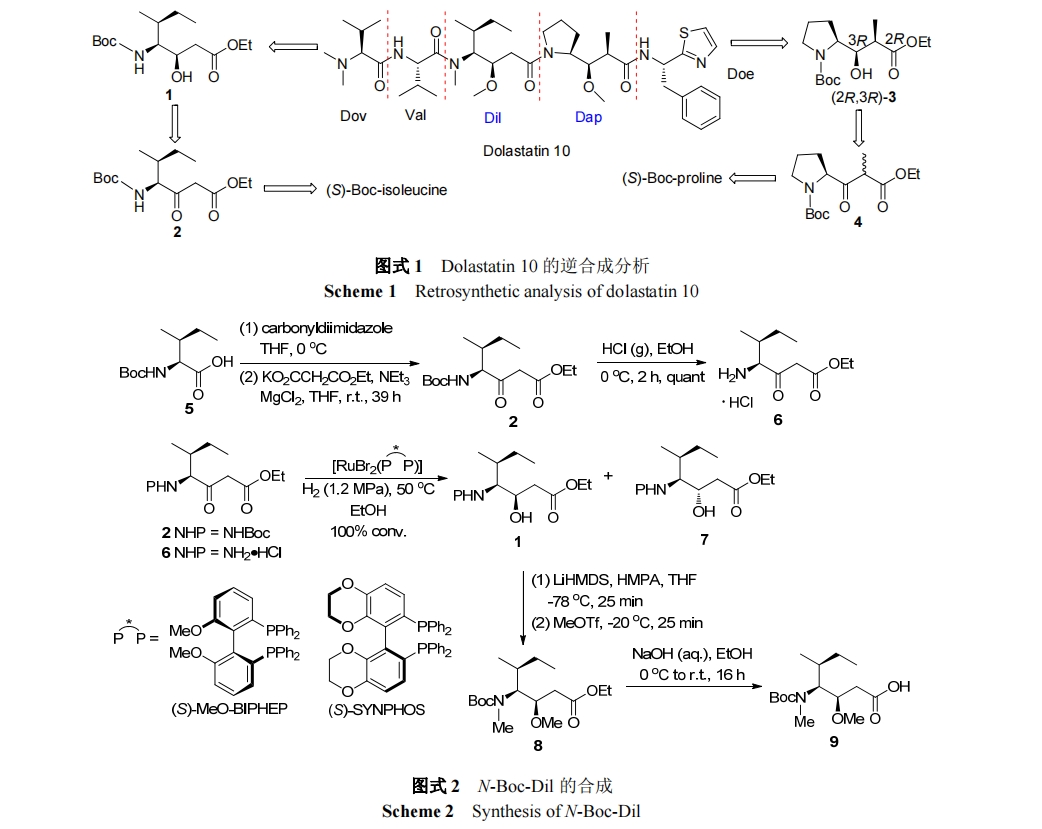
1.1 Dolastatin 10
Dolastatin 10 «“Μ÷÷œΏ–‘ΈεκΡ, ΑϋΚ§ΈεΗωΉ”ΒΞ‘Σ, dolavaline (Dov), valine (Val), dolaisoleuine (Dil), dolaproine (Dap)ΚΆ dolaphemine (Doe)[15]. Mordant Β»[16]Ά®ΙΐΡφΚœ≥…Ζ÷ΈωΖΔœ÷DapΚΆDil «Κœ≥…ΒΡΝΫΗωΙΊΦϋΤ§ΕΈ(Scheme 1), “ρ¥ΥΥϊΟ«œ»Κœ≥…ΝΥ N-Boc-Dil (9) (Scheme 2)ΚΆN-Boc- (2R,3R)-Dap (11) (Scheme 3). ‘Ό“‘ 9 ΚΆ 11 ΈΣ‘≠ΝœΚœ≥…Dolastatin 10 (Scheme 4), ‘ΎΚœ≥…Ιΐ≥Χ÷–ΫΙΧΩΥαΕΰ““θΞ(DEPC)ΚΆ»ΐΖζ““Υα(TFC)÷ς“Σ”Ο”Ύ≈ΦΝΣΚΆΆ―±ΘΜΛ. Dolastatin 10Ής”ΟΜζ÷Τ÷ς“Σ «Ά®Ιΐ”κΠ¬ΈΔΙήΒΑΑΉΫαΚœ, ΙœΗΑϊΖ÷Ν―ΆΘ÷Ά, Ήν÷’ΒΦ÷¬œΗΑϊΥάΆω. ‘ΎΡ≥–©ΝΌ¥≤«Α ‘―ι÷–, Dolastatin 10 Ε‘“Μ–© ΒΧεΝω±μœ÷≥ωΝΥΫœΚΟΜν–‘, »γΕ‘ΥΡ÷÷Ζ«–ΓœΗΑϊΖΈΑ© (NCI-H69, NCI-H82, NCI-H446, NCI-H510) IC50÷ΒΖΕΈßΈΣ 0.032ΓΪ0.184 nmol•LΘ≠1[17].
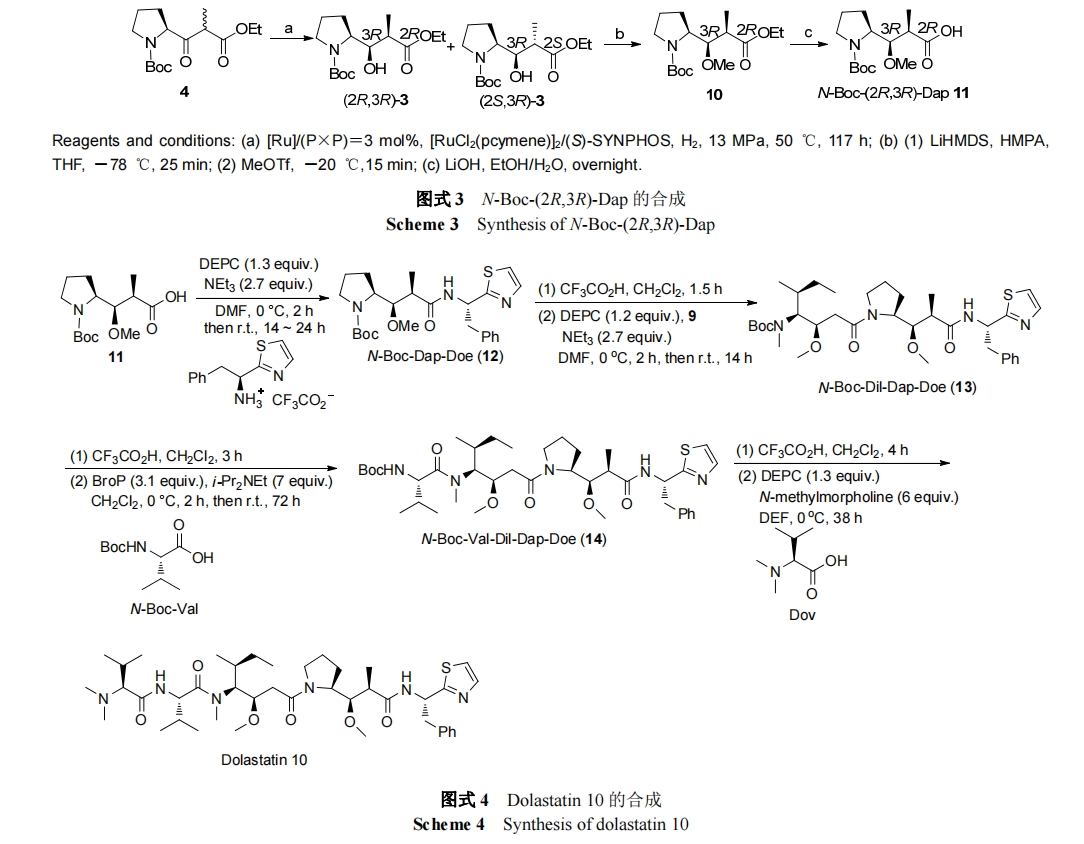
Perez Β»[18]ΚΆ Kindler Β»[19]Ε‘ Dolastatin 10 ÷ΈΝΤΆμΤΎ»ιœΌΑ©ΚΆΆμΤΎ“»Β®Α©ΩΣ’ΙΝΥ II ΤΎΝΌ¥≤ ‘―ι. Υδ»Μ, Dolastatin 10 ‘ΎΧεΆβ ‘―ι–ßΙϊΫœΚΟ, ΒΪΝΌ¥≤ ‘―ιΫαΙϊ±μΟς, Β―ιΉιΜΦ’ΏΒΡ÷–ΈΜ…ζ¥φΤΎΫœΕΧ, ÷ΈΝΤ–ßΙϊ≤ΜΦ―. ΥφΚσ, Pettit ΚΆ Miyazaki Β»ΈΣΝΥΦλ―ιΫαΙΙΗΡ±δΕ‘Μν–‘ΒΡ”Αœλ, Ε‘ Dolastatin 10 ΒΡ C ΕΥΜρ N ΕΥΫχ–––ό Έ, Κœ≥…ΝΥ“ΜœΒΝ–Dolastatin 10 ΒΡ―ή…ζΈο, ΟϋΟϊΈΣ auristatins œΒΝ–, »γauristatin E, monomethyl auristatin D (MMAD) ΚΆmonomethyl auristatin E Β»Μ·ΚœΈο(ΆΦ 1), ΒΪ MMAD ΒΞ“©÷ΈΝΤΒΡΧεΡΎ Β―ι±μΟςΤδΩΙ÷ΉΝωΜν–‘≤ΜΦ―. ÷°Κσ, SenterΒ»ΖΔœ÷ Auristatins ΒΡ N ΕΥ÷ΌΑΖΩ…Φ”‘ΊΝ§Ϋ”Χε“‘≈ΦΚœΒΞΩΥ¬ΓΩΙΧε, –Έ≥…ΗΏ–ßΓΔΗΏΑ–œρ–‘ΒΡΩΙΧε-“©Έο≈ΦΚœΈο[20].
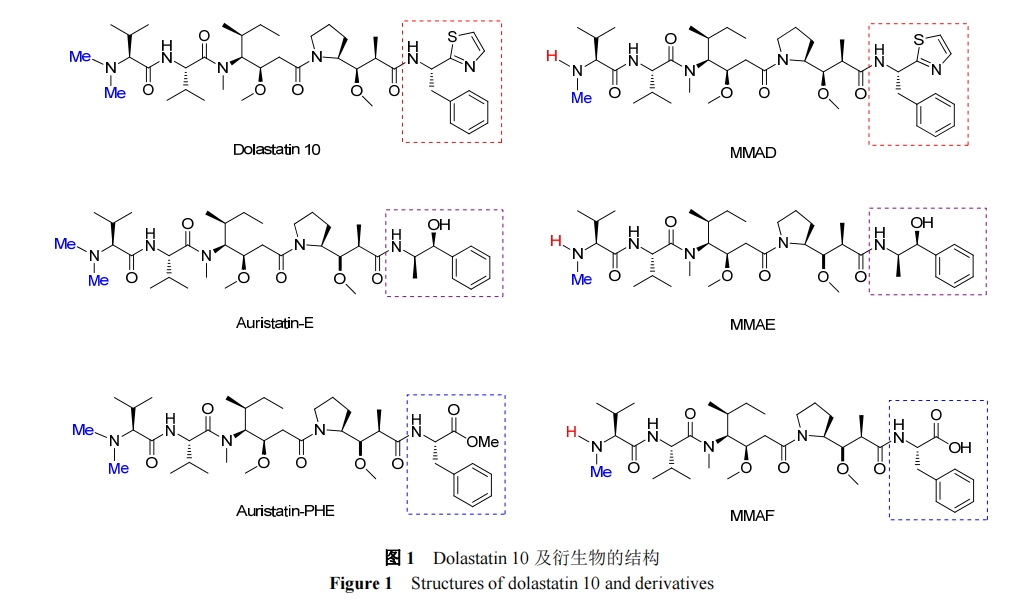
1.2 Μυ”Ύ auristatins ΒΡΩΙΧε-“©Έο≈ΦΚœΈο
ΩΙΧε-“©Έο≈ΦΚœΈο(ADC) «ΨΏ”–ΗΏΜν–‘ΓΔΗΏΑ–œρ–‘ΒΡ–¬–ΥΩΙΑ©÷ΈΝΤ ‘ΦΝ. Υϋ”…ΩΙΧεΓΔΝ§Ϋ”ΧεΓΔœΗΑϊΕΨ–‘ ‘ΦΝ»ΐ≤ΩΖ÷ΙΙ≥…. œΗΑϊΕΨ–‘ ‘ΦΝΒΡ–ßΝΠΓΔΑ–œρΒΡ―Γ‘ώΚΆΝ§Ϋ”ΧεΒΡΈ»Ε®–‘Β» «”Αœλ–¬–Ά ADC ΒΡ“ρΥΊ. ΡΩ«Α, Ν§Ϋ”ΧεΦΦ θΒΡΖΔ’ΙΚΆ–¬–ΆΗΏ–ßΒΡœΗΑϊΕΨ–‘ ‘ΦΝΒΡ≥ωœ÷ΕΦΦΪ¥σΒΊ¥ΌΫχΝΥ ADC ΒΡΖΔ’Ι[21]. ‘ΎΚΘ―σ…ζΈοΜν–‘κΡ÷–, “‘Dolastatin 10 ΈΣΡΗΧεΜ·ΚœΈοΚœ≥…ΒΡ auristatins œΒΝ–±ΜΙψΖΚ”Π”Ο”Ύ ADC[22]. œ÷‘Φ”– 45 ÷÷ Auristatins άύΒΡ ADC ¥Π”ΎΝΌ¥≤―–ΨΩΫΉΕΈ[23]. œ¬ΟφΫΪ“‘≤¥» ΆΉΈςΖ≤ΕύΆΓ, Depatuxizumab mafodotin, Glembatumumab Vedotin, Polatuzumabvedotin ΦΑ AGS-67E ΈΣάΐΫι…ή auristatins άύADC.
1.2.1 ≤¥» ΆΉΈςΖ≤ΕύΆΓ
≤°» ΆΉΈςΖ≤ΕύΆΓ(Brentuximab vedotin, SGN-35)”…»’±ΨΈδΧο(Takeda)÷Τ“©ΙΪΥΨΦΑΟάΙζΈς―≈ΆΦ“≈¥Ϊ―ßΙΪΥΨ(Seattle Genetics)ΝΣΚœ―–ΖΔ, ”Ύ 2011 Ρξ”…ΟάΙζ ≥ΤΖ“©ΤΖΙήάμΨ÷ (FDA) ≈ζΉΦ…œ – , ”Ο”Ύ÷ΈΝΤΜτΤφΫπΝήΑΆΝω(HodgkinΓ·s lymphoma, HL)ΚΆœΒΆ≥–‘Φδ±δ–‘¥σœΗΑϊΝήΑΆΝω(Anaplastic large cell lymphoma, ALCL)[24].
≤°» ΆΉΈςΖ≤ΕύΆΓΩ…”κ±μ¥ο CD30 ΒΡ÷ΉΝωœΗΑϊΗΏΕ»ΧΊ“λ–‘ΫαΚœ, Υϋ”… cAC10 (CD30 ΩΙΧε)ΓΔΝ§Ϋ”ΧεΚΆœΗΑϊΕΨ ‘ΦΝmonomethyl auristatin E (MMAE)»ΐ≤ΩΖ÷Ήι≥…. ≤°» ΆΉΈςΖ≤ΕύΆΓΥυ Ι”ΟΒΡΝ§Ϋ”ΧεΈΣγ”Α±Υα-ΙœΑ±Υα(vc), ΈΣ±ψ”ΎΝ§Ϋ”Χε(vc)ΚΆΕΨ–‘ ‘ΦΝΥ°Ϋβ«“ ΙΥ°Ϋβ≤ΩΈΜΚΆœΗΑϊΕΨ ‘ΦΝΒΡΜν–‘≤ΩΈΜ‘Εάκ, Ι ”ΟΕ‘Α±Μυή–―θτ Μυ(PABC)ΉςΈΣΦδΗτΧεΦϋΚœ”Ύ MMAE ”κΝ§Ϋ”Χε÷°Φδ , –Έ ≥…vc-PABC-MMAE ΫαΙΙ, ‘ΌΫΪΤδΆ®Ιΐ¬μά¥θΘ―«ΑΖΦΚθΘΜυ(mc)”κΒΞΩΥ¬ΓΩΙΧε÷–ΒΡΑκκΉΑ±ΥαΝ§Ϋ”[25]. CD30 «ΡΛ…œΒΡΧ«ΒΑΑΉ, τ”Ύ÷ΉΝωΜΒΥά“ρΉ”(TNF) ήΧεΦ“Ήε≥…‘±, Υϋ‘Ύ HL, ALCL Β»Εύ÷÷ΝήΑΆΝω±μΟφΗΏΕ»±μ¥ο. cAC10 ±Ψ…μΨΏ”–Ε‘ΩΙ CD30 ΒΡΜν–‘, ΒΪ‘ΎΝΌ¥≤ II ΤΎ ±Μν–‘œϊ ß, ΈΣ‘ω«ΩΤδΜν–‘, Κœ≥…ΝΥ≤°» ΆΉΈςΖ≤ΕύΆΓ. œΗΑϊΕΨ ‘ΦΝMMAE «¥””ΓΕ»―σΈόΩ«»μΧεΕ·ΈοΫΊΈ≤ΚΘΆΟ Dolabella auriculariaΖ÷άκΒΟΒΫDolastatin 10ΒΡΚœ≥…―ή…ζΈο. ‘Ύ…ζάμΧθΦΰœ¬, MMAE ΨΏ”–ΗΏ–ßΓΔ»ήΫβΕ»ΗΏΓΔΈ»Ε®–‘ΚΟΒ»”≈Βψ. MMAE ΒΡ“©άμΉς”Ο”κ Dolastatin 10 œύΥΤ, Ά®Ιΐ“÷÷ΤΈΔΙήΒΑΑΉΒΡΨέΚœ, Ι±μ¥ο CD30 ΒΡΝήΑΆΝωœΗΑϊ‘Ύ G2-M ΫΉΕΈ‘ω≥ΛΆΘ÷Ά, ¥”Εχ ΙœΗΑϊΒρΆω. ≤¥» ΆΉΈςΖ≤ΕύΆΓΉς”ΟΜζ÷Τ «Ά®Ιΐ cAC10 Α–œρ Ε±π÷ΉΝωœΗΑϊ…œΒΡ CD30, ”κ CD30ΫαΚœ, »ΜΚσΨ≠œΗΑϊΆχΗώΒΑΑΉΒΡΒςΫΎ, ≤¥» ΆΉΈςΖ≤ΕύΆΓ±ΜÉ»ΆΧΫχ»κœΗΑϊ, ‘Ύ»ήΟΗΧε÷–±ΜΒΑΑΉΟΗΥ°Ϋβ ΆΖ≈ MMAE, MMAE ‘ΎœΗΑϊΡΎΖΔΜ”Ής”Ο , …±ΥάœΗΑϊ [26]. Mc-vc-PBAC-MMAE ΒΡΚœ≥… «Ά®Ιΐ”Ο N-ΦΉΜυγ”Α±ΥαάύΜ·ΚœΈο 15 ¥ζΧφ N,N-ΕΰΦΉΜυγ”Α±ΥαάύΜ·ΚœΈοά¥±ΘΜΛauristatain E, ¥”Εχ–Έ≥… MMAE (18). ”Ο mc-vc-PBAC (19)Ϋχ“Μ≤Ϋ–ό ΈΜ·ΚœΈο 18 ΒΟΒΫ mc-vc-PBAC-MMAE (20), ΉνΚσ ΙΜΙ‘≠ΒΡcAC10”κΜ·ΚœΈο20ΫαΚœΦ¥Ω…ΒΟΒΫ≤°» ΆΉΈςΖ≤ΕύΆΓ(Scheme 5)[27].
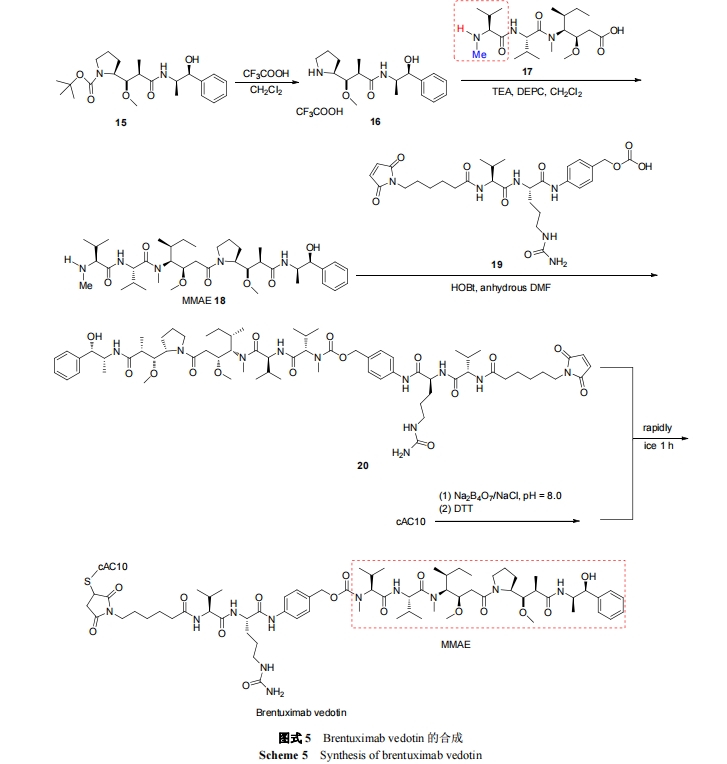
ΈΣΤάΦέ Brentuxmab vedotin Ε‘Ή‘…μΗ…œΗΑϊ“Τ÷≤ΚσΜρΝΫ¥ΈΦ»ΆυΜ·ΝΤΚσΦ≤≤Γ”–Ϋχ’Ι≤ΜΡήΫ” ή“Τ÷≤ΒΡ HL ΜΦ’ΏΚΆ1 ¥ΈΦ»ΆυΜ·ΝΤΚσΦ≤≤ΓΫχ’Ι ALCL ΜΦ’ΏΒΡ÷ΈΝΤ–ßΙϊ, ‘Ύ±±ΟάΚΆ≈Ζ÷όΒΡ 78 ΗωΒΊΒψΫχ––ΝΥ III ΤΎΝΌ¥≤ ‘―ι. ‘―ι―Γ‘ώΝΥ 329 άΐΖϊΚœ“Σ«σΒΡΜΦ’ΏΥφΜζΖ÷≈δ÷Ν Brentuxmab vedotin Ήι(nΘΫ165ΚΆΑ≤ΈΩΦΝΉι(nΘΫ164). ΫαΙϊœ‘ Ψ, ”κΑ≤ΈΩΦΝΉιœύ±», Brentuxmab vedotin ΉιΒΡΜΦ’ΏΈόΫχ’Ι…ζ¥φΤΎΒΟΒΫœ‘÷χΗΡ…Τ([HR] 0.57, 95% CI 0.40ΓΪ0.81; pΘΫ0.0013), Brentuxmab vedotin ΉιΜΦ’Ώ, ÷–ΈΜΈόΫχ’Ι…ζ¥φΤΎΤΫΨυ‘ΦΈΣ 42.9 Ηω‘¬(95% CI 30.4ΓΪ42.9), Α≤ΈΩΦΝΉιΒΡΜΦ’Ώ, ÷–ΈΜΈόΫχ’Ι…ζ¥φΤΎΤΫΨυ‘ΦΈΣ 24.1 Ηω‘¬(11.5ΓΪnot estimabl). III ΤΎΝΌ¥≤Α≤»Ϊ–‘ ‘―ιΆ≥ΦΤ 167 Οϊ Brentuximab vedotin ΉιΜΦ’ΏΚΆ 160 ΟϊΑ≤ΈΩΦΝΉιΜΦ’ΏΒΡ≤ΜΝΦΖ¥”Π(Adversedrug reaction, ADR)ΖΔ…ζ«ιΩω, ΝΫΉι ADR ΖΔ…ζ¬ Ζ÷±πΈΣ 98%ΚΆ 89%, ―œ÷Ί≤ΜΝΦΖ¥”Π¬ (ΓίGrade 3)Ζ÷±πΈΣ 56%ΚΆ 32%. ‘Ύ Brentuximab vedotin Ήι÷–, Ήν≥ΘΦϊΒΡ≤ΜΝΦΖ¥”Π «Άβ÷ήΗ–Ψθ…ώΨ≠≤Γ±δ, ΖΔ…ζ¬ ΈΣ 30.5%; “ρΆβ÷ή…ώΨ≠≤Γ±δΒΦ÷¬ 51 »ΥΆΘ÷Ι Ι”Ο Brentuximab vedotin ÷ΈΝΤ. ΤδΥϋ≥ΘΦϊΒΡ ADR ”–÷––‘ΝΘœΗΑϊΦθ…ΌΓΔ…œΚτΈϋΒάΗ–»ΨΓΔΤΘάΆΓΔΆβ÷ή‘ΥΕ·…ώΨ≠≤ΓΓΔΕώ–ΡΓΔΩ»Υ‘ΓΔΗΙ–ΚΓΔΖΔ»»ΓΔ≈ΜΆ¬Β»[28].
1.2.2 Depatuxizumab mafodotin (ABT-414)
Depatuxizumab mafodotin Α–œρΉς”Ο”Ύ±μΤΛ“ρΉ” ήΧε(EGFR), ”…ΩΙΧε(ABT-806)ΓΔΝ§Ϋ”ΧεΚΆœΗΑϊΕΨ ‘ΦΝmonomethyl auristatin F (MMAF) ΙΙ ≥… ( ΆΦ 2)[29]. ‘ΎDepatuxizumab mafodotin ÷–, Ι”ΟΒΡΝ§Ϋ”Χε «Έ»Ε®–‘ΫœΗΏΒΡ¬μά¥θΘ―«ΑΖΦΚθΘΜυ(mc). FDA ΚΆ≈Ζ÷ό“©ΈοΙήάμΨ÷(EMA)”Ύ 2014 Ρξ≈ζΉΦ Depatuxizumab mafodotin ΉςΈΣ÷ΈΝΤΫΚ÷ œΗΑϊΝωΒΡΙ¬Ευ“©[30].
EGFR «±μΤΛ…ζ≥Λ“ρΉ” ήΧε(HER)Φ“Ήε≥…‘±÷°“Μ, ”κœΗΑϊΒΡ…ζ≥ΛΓΔ‘ω÷≥ΚΆΖ÷Μ·”–ΙΊ. EGFR ‘Ύ–μΕύ÷ΉΝωœΗΑϊ±μΟφ¥φ‘ΎΗΏ±μ¥οΜρ“λ≥Θ±μ¥οœ÷œσ, ―–ΨΩΖΔœ÷Υϋ”κ÷ΉΝωœΗΒΡ‘ω÷≥ΓΔ―ΣΙή…ζ≥…ΓΔ÷ΉΝω«÷œ°ΓΔΉΣ“ΤΦΑœΗΑϊΒρΆωΒΡ“÷÷Τ”–ΙΊ. –Γ σΩΙΧε mAb 806 «“‘ EGFRvIII(»±…ΌΆβœ‘Ή”2ΓΪ7 ΒΡ≈δΧεΫαΙΙ”ρ≤Δ±ΘΝτΉι≥…–ΆΦΛΟΗΜν–‘ΒΡ EGFR ΒΡΉν≥ΘΦϊ»± ßΆΜ±δΧε)ΜρΙΐ±μ¥οΒΡ‘≠…ζ–Ά EGFR ΈΣΑ–±ξ, ΈΣΫΒΒΆ σ‘¥ΩΙΧεΩ…Ρή“ΐΖΔΒΡΙΐΟτΖ¥”Π≤Δ Βœ÷ΝΌ¥≤ΩΣΖΔ,Ε‘ mAb 806 Ϋχ––»Υ‘¥Μ·ΒΟΒΫ÷ΊΉι IgG1/k mAb ABT-806. ABT-806 Ε‘ EGFRv III ΜρΙΐ±μ¥οΒΡ EGFR ΫαΚœΝΠΗϋ«Ω, ΧΊ“λ–‘ΗϋΗΏ, ΫΒΒΆ’ΐ≥ΘΉι÷·ΒΡ ADR ΖΔ…ζ¬ [31]. MMAFΆ§―υ « Dolastatin 10 ΒΡΚœ≥…―ή…ζΈο, Doronina Β»[32]±®ΒάΝΥ MMAF ΒΡΙΧœύΚœ≥…¬ΖœΏ, “‘ Phe-2-¬»»ΐ±ΫΦΉΜυ ς÷§21 ΈΣ‘≠Νœ, Ψ≠Ιΐ≈ΦΝΣΆ―±ΘΜΛΒΟΒΫ MMAF. MeVal-ValDil-Dap-Phe-2-¬»»ΐ±ΫΦΉΜυ ς÷§»τ÷±Ϋ””κ mc ≈ΦΝΣ, Ά―Βτ¬»»ΐ±ΫΦΉΜυ ς÷§‘ρΒΟΒΫ mcMMAF (Scheme 6).
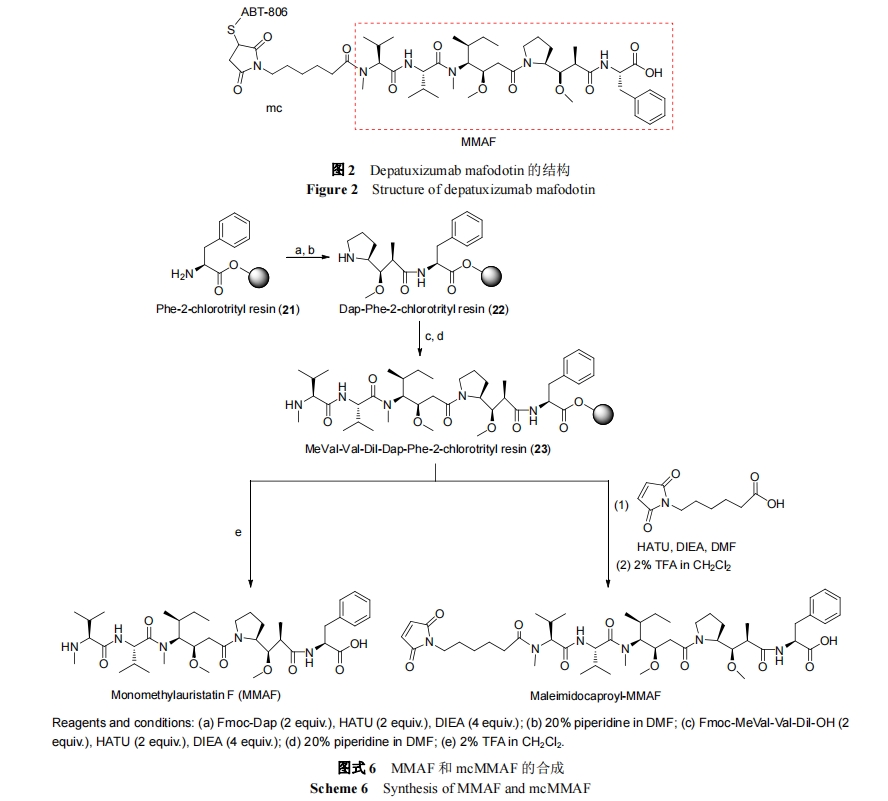
ΈΣΤάΙά ABT-414 ”κΧφΡΣΏρΑΖΝΣΚœ÷ΈΝΤΗ¥ΖΔ–‘ΜρΡ―“‘«–≥ΐΕώ–‘ΫΚ÷ ΝωΒΡΑ≤»Ϊ–‘ΚΆ”––ß–‘, Ε‘ 12 ΈΜ ή ‘’ΏΩΣ’ΙΝΥ I ΤΎΝΌ¥≤ ‘―ι. 9 ΈΜΜΦ’Ώ÷ΈΝΤ–ßΙϊΫœΚΟ, Τδ÷– 1 ΈΜΆξ»ΪΜΚΫβ, 2 ΈΜ≤ΩΖ÷ΜΚΫβ. ΖΔœ÷“ΜΑψ ADR (Γί3 ΟϊΜΦ’Ώ)Αϋά® ”ΝΠΡΘΚΐ(nΘΫ5), Ϋ«ΡΛ≥ΝΜΐ(nΘΫ4)ΓΔ―έ≤Ω“λΈοΗ–ΓΔΕώ–ΡΓΔΖΔ»»ΚΆΆΖΆ¥(ΟΩœν nΘΫ3). ―œ÷Ί ADR Αϋά®ΝήΑΆ«ρΦθ…Ό÷ΔΓΔΫ«ΡΛ≥ΝΜΐΓΔΤΛΖτΗ–»ΨΚΆ―Σ“ΚΒ®ΙΧ¥Φ…ΐΗΏ(ΟΩœν nΘΫ1). ‘Ύ“ΜΕ®ΦΝΝΩΖΕΈßΡΎ(0.5ΓΪ1.0 mg/kg)“©¥ζΕ·ΝΠ―ß(PK)≤Έ ΐ”κΦΝΝΩ≥…±»άΐ, ΑκΥΞΤΎΈΣ 7ΓΪ8 d. ΤδΫαΙϊ±μΟςABT-414 Ε‘”Ύ÷ΈΝΤΗ¥ΖΔ–‘ΜρΡ―“‘«–≥ΐΕώ–‘ΫΚ÷ Νω÷ΒΒΟΫχ“Μ≤ΫΒΡ―–ΨΩ[33].
1.2.3 Glembatumumab Vedotin (CDX-011)
Glembatumumab Vedotin «”… CuraGen ΙΪΥΨ―–ΖΔΒΡADC, ΡΩ«Α¥Π”Ύ÷ΈΝΤΉΣ“Τ–‘»ιœΌΑ©ΓΔΚΎ…ΪΥΊΝωΓΔΙ«»βΝωΒΡΝΌ¥≤ II ΤΎ ‘―ι“‘ΦΑ÷ΈΝΤΝέΉ¥œΗΑϊΑ©ΒΡΝΌ¥≤ I/II ΤΎ ‘―ιΫΉΕΈ. 2010 Ρξ FDA ΫΪ Glembatumumab Vedotin Φ”»κΩλΥΌ…σ≈ζΟϊΒΞ, “‘÷ΈΝΤΆμΤΎΓΔΆγΙΧ–‘ΓΔΗΏ±μ¥οΖ«ΉΣ“Τ–‘ΚΎ…ΪΥΊΝωΧ«ΒΑΑΉ B (GPNMB)»ιœΌΑ©.
Glembatumumab Vedotin Α–œρΉς”Ο”Ύ GPNMB, ”…ΩΙΧε CR011(“‘ GPNMB ΈΣΑ–ΒψΒΡ»Υ‘¥Μ· IgG2ΒΞΩΥ¬ΓΩΙΧε)ΓΔΝ§Ϋ”ΧεΚΆ MMAE ΙΙ≥…(ΆΦ 3). GPNMB «“Μ÷÷ΩγΡΛΧ«ΒΑΑΉ, ‘ΎΕύ÷÷’ΐ≥ΘΉι÷·÷–Ψυ”–±μ¥ο, »γΙ«ςάœΒΆ≥ΓΔ‘λ―ΣœΒΆ≥ΓΔΤΛΖτΒ», ”κ≥…Ι«–Έ≥…ΚΆΚΎ…ΪΥΊœΗΑϊ…ζ≥…”–ΙΊ; ΒΪ GPNMB ΗΏΕ»±μ¥ο”Ύ»ιœΌΑ©ΓΔΚΎ…ΪΥΊΝωΓΔΙ«»βΝωΒ»Εύ÷÷Εώ–‘÷ΉΝω÷–, ‘ΎΑ©÷ΔΉΣ“ΤΓΔ÷ΉΝωœΗΑϊ«÷œ°ΚΆ«®“Τ÷–ΖΔΜ”÷Ί“ΣΉς”Ο. Glembatumumab Vedotin ΒΡΉς”ΟΜζ÷Τ”κBrentuximab vedotin œύΥΤ, Ήœ»Ά®ΙΐΩΙΧε CR011 Ε±πΑ–Βψ, ΙΤδ”κ GPNMB –Έ≥…Η¥ΚœΈοΫχ»κœΗΑϊ, ‘Ό‘Ύ»ήΟΗΧε÷–±ΜΒΑΑΉΟΗΥ°Ϋβ, ΆΖ≈ MMAE, MMAE Ά®Ιΐ“÷÷ΤΈΔΙήΒΑΑΉΨέΚœ, Ι”–ΥΩΖ÷Ν―ΆΘ÷Ά, ΒΦ÷¬œΗΑϊΒρΆω[34].
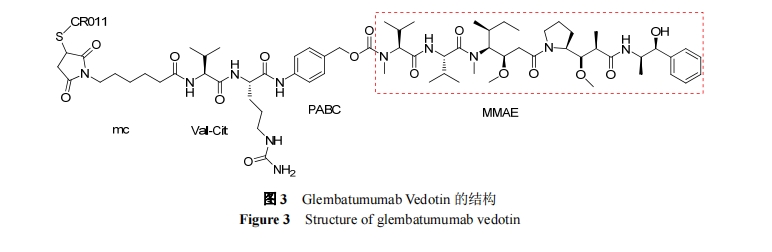
ΈΣΤάΙά Glembatumumab Vedotin ÷ΈΝΤΆμΤΎΓΔΆγΙΧ–‘ΓΔΗΏ±μ¥ο GPNMB »ιœΌΑ©ΒΡΑ≤»Ϊ–‘ΚΆ”––ß–‘, Ε‘ 124 ΟϊΜΦ’ΏΫχ––ΝΥ II ΤΎΝΌ¥≤ ‘―ι. ΫΪΥυ”– ή ‘’Ώ“‘ 2ΓΟ1 ΒΡ±»άΐΥφΜζΖ÷≥…ΝΫΉι, “ΜΉιΈΣ Glembatumumab Vedotin Ήι(nΘΫ83), Νμ“ΜΉι”…―–ΨΩ’Ώ―Γ‘ώ(IC)“©Έο(Α§»’≤ΦΝ÷ΓΔΩ®≈ύΥϊ±θΓΔ≥Λ¥Κ»π±θΓΔΦΣΈςΥϊ±θΓΔΉœ…Φ¥ΦΓΔΉœ…άΧΊΕϊΓΔ―ΈΥαΕύ»α±»–«ΓΔΕύ»α±»–«÷§÷ ΧεΓΔΉœ…Φ¥Φ”κΑΉΒΑΑΉΫαΚœ–ΆΜλ–ϋ“Κ), ΒΞ“©Μ·ΝΤ(nΘΫ41). ΤδΫαΙϊ±μΟς, ”κ IC “©Έοœύ±», Glembatumumab Vedotin ±μœ÷≥ωΫœΚΟΒΡΡΆ ή–‘, ΫœΒΆΒΡ―Σ“ΚΕΨ–‘; ≥ωœ÷ΒΡ“ΜΑψ ADR ”–ΤΛ’νΓΔπΰ―ςΓΔ…ώΨ≠≤ΓΚΆΆ―ΖΔ. ‘Ύ≥θΦΕ÷’Βψ, Glembatumumab Vedotin Ήι”κ IC Μ·ΝΤΉιœύ±», ΩΆΙέΜΚΫβ¬ ΟΜ”–œ‘÷χ–‘≤ν“λ. ΒΪ‘Ύ¥ΈΦΕ÷’Βψ, Glembatumumab Vedotin Ήι÷–ΗΏ±μ¥ο GPNMB (Γί25%ΒΡ÷ΉΝωœΗΑϊ)ΜΦ’ΏΒΡΩΆΙέΜΚΫβ¬ (ORR)ΈΣ 30%, Οςœ‘ΗΏ”ΎIC Μ·ΝΤΉιΒΡ 9%. ‘Ύ¥Υ¥Έ ‘―ι÷–, ΜΙ≤βΒΟΝΥΜΦ”–»ΐ“θ–‘»ιœΌΑ©ΜΦ’ΏΒΡ ORR, Glembatumumab Vedotin ΉιΈΣ 18%, IC Μ·ΝΤΉιΈΣ 0%. Ε‘”ΎΙΐΕ»±μ¥ο GPNMB ΒΡ»ΐ“θ–‘»ιœΌΑ©ΜΦ’Ώ, Glembatumumab Vedotin Ήι ORR ΈΣ 40%, IC Μ·ΝΤΉιΈΣ 0%. ¥ΥΫαΙϊ±μΟς, Glembatumumab Vedotin Ε‘”Ύ÷ΈΝΤΙΐΕ»±μ¥οGPNMBΒΡ»ιœΌΑ©ΜΦ’Ώ, ΨΏ”–ΫœΚΟΒΡΝΤ–ß, ”»ΤδΕ‘”Ύ»ΐ“θ–‘»ιœΌΑ©’Ιœ÷≥ωΫœΗΏΒΡΜν–‘[35].
1.2.4 Polatuzumab vedotin (RG-7596)
Polatuzumab vedotin «Α–œρΉς”Ο”Ύ CD79b ΒΡ ADC, ”…»Υ‘¥Μ·ΒΡΩΙΧεΓΔΝ§Ϋ”ΧεΚΆ MMAE ΙΙ≥…. œ÷‘Ύ¥Π”Ύ÷ΈΝΤΟ÷¬ΰ¥σ B œΗΑϊΝήΑΆΝωΚΆΤδΥϊ÷÷άύΖ«ΜτΤφΫπΝήΑΆΝωΒΡΝΌ¥≤ II ΤΎ―–ΨΩΓΔ÷ΈΝΤ¬Υ≈ί–‘ΝήΑΆΝωΒΡΝΌ¥≤ I/II ΤΎ ‘―ι“‘ΦΑ÷ΈΝΤ¬ΐ–‘ΝήΑΆœΗΑϊΑΉ―Σ≤ΓΒΡΝΌ¥≤ I ΤΎ―–ΨΩΫΉΕΈ. CD79b ‘Ύ B œΗΑϊΓΔ¬ΐ–‘ΝήΑΆœΗΑϊΑΉ―Σ≤Γ(CLL)ΓΔB œΗΑϊΖ«ΜτΤφΫπΝήΑΆΝω(B-NHLs)÷–±μ¥ο, «÷ΈΝΤΖ«ΜτΤφΫπ œΝήΑΆΝωΒΡΝΦΚΟΑ–±ξ. Polatuzumab vedotin Υυ Ι”ΟΒΡΝ§Ϋ”ΧεΓΔœΗΑϊΕΨ ‘ΦΝ”κ Brentuximab vedotin œύΆ§, ΈΣ mc-vcPABC-MMAE(ΆΦ 4, ΩΙΧεΈΣ»Υ‘¥Μ·ΩΙΧε)[36].
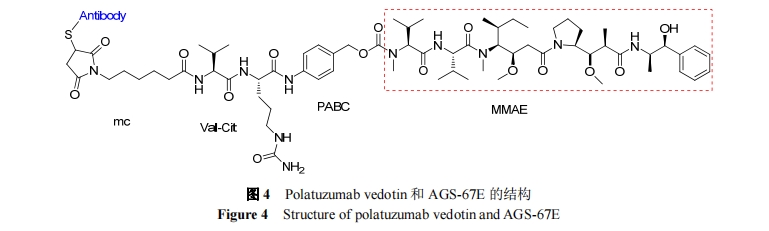
ΈΣΫχ“Μ≤ΫΤάΙά Polatuzumab vedotin ÷ΈΝΤ NHL ΚΆCLL ΒΡΑ≤»Ϊ–‘ΦΑ”––ß–‘, Palanca-Wessels ΦΑΤδΆ§ ¬[37]Ρ…»κ 95 άΐΜΦ’ΏΫχ–– II ΤΎΝΌ¥≤ ‘―ι, ’β–©ΜΦ’ΏΨ≠Ιΐ I ΤΎΓΔΕύ÷––ΡΓΔΖ«ΟΛ―–ΨΩ, ±μ¥ο CD79B, Ρ―“‘÷ΈΝΤ«“÷ΈΝΤ“β“ε≤Μ¥σ. ‘―ιΉι±π…η÷ΟΈΣ: NHL ΦΝΝΩΒί‘ωΉι 34 Οϊ, CLL ΦΝΝΩΒί‘ωΉι18Οϊ, “‘»ΖΕ®Polatuzumab vedotinΒΡΉν¥σΡΆ ήΦΝΝΩ; IIΤΎΆΤΦωΦΝΝΩ NHLά©’ΙΉι 34 Οϊ, NHL άϊΆΉΈτΒΞΩΙΝΣΚœΉι9Οϊ(”…”ΎΦΝΝΩΒί‘ωΉι»±ΖΠΝΤ–ßΈ¥…η÷ΟCLLά©’ΙΉι)NHL ΜΦ’ΏΉν≥ΘΦϊΒΡ ADR ΈΣ÷––‘ΝΘœΗΑϊΦθ…Ό÷Δ, ΤΕ―ΣΚΆ÷ήΈß…ώΨ≠≤Γ±δ. ‘Ύ 42 ΟϊΩ…ΤάΙάΒΡ NHL ΜΦ’Ώ÷–, 23 ΟϊΩΆΙέΜΚΫβ, Τδ÷– 7 ΟϊΆξ»ΪΜΚΫβΓΔ16 Οϊ≤ΩΖ÷ΜΚΫβ. Polatuzumab vedotin ΚΆάϊΆΉΈτΒΞΩΙΝΣΚœ÷ΈΝΤΒΡ 9 ΟϊΜΦ’Ώ÷–7 ΟϊΜΦ’ΏΩΆΙέΜΚΫβ, Αϋά® 2 ΟϊΆξ»ΪΜΚΫβΚΆ 5 Οϊ≤ΩΖ÷ΜΚΫβ. ΝΌ¥≤ ‘―ιΫαΙϊ±μΟς, Polatuzumab vedotin ÷ΈΝΤ NHL ΜΦ’ΏΑ≤»Ϊ–‘ΚΆΡΆ ή–‘ΝΦΚΟ, ΒΪΕ‘ CLL ΜΦ’Ώ‘ρΈό–ß, “ρ¥Υ’β“ΜΝΤ–ß”Π‘Ύ NHL ΖΫΟφΫχ––Ϋχ“Μ≤ΫΤάΙά.
1.2.5 AGS-67E
AGS-67E «”… Agensys ΙΪΥΨ(Α≤ΥΙΧ©ά¥ΒΡΉ”ΙΪΥΨ)―–ΖΔ”Ο”Ύ÷ΈΝΤΦ±–‘Υη–‘ΑΉ―Σ≤Γ(AML)ΓΔΝήΑΆœΗΑϊΑΉ―Σ≤ΓΒ»―Σ“ΚΕώ–‘÷ΉΝωΒΡ ADC. œ÷¥Π”ΎΝΌ¥≤ I ΤΎΒΡ―–ΨΩΫΉΕΈ.
AGS-67E Α–œρ”Ύ CD37, ”…Άξ»Ϊ»Υ‘¥Μ· IgG2–ΆΒΞΩΥ¬ΓΩΙΧεΓΔΝ§Ϋ”ΧεΚΆMMAEΙΙ≥…(ΆΦ4, ΩΙΧεΈΣ»Υ‘¥Μ·IgG2ΩΙΧε). CD37 «ΥΡΩγΡΛΒΑΑΉ≥§Φ“Ήε(TM4SF)ΒΡ≥…‘±, ΨΏ”–ΥΡΗω«±‘ΎΒΡΩγΡΛ«χ. CD37 Ά®Ιΐ C ΚΆ N Ρ©ΕΥΫαΙΙ”ρΩ…“‘÷±Ϋ”ΫιΒΦΥΪ–≈Κ≈ΉΣΒΦ. CD37 ‘Ύ”…‘≠ Φ B œΗΑϊΒΫΆβ÷ή≥… λ B œΗΑϊΒΡΖΔ”ΐΙΐ≥Χ÷–±μ¥οΝΩ÷πΫΞ‘ωΦ”, ΒΪ‘ΎΫ§œΗΑϊΓΔT œΗΑϊΦΑΒΞΚΥœΗΑϊ÷–≤Μ±μ¥ο, ‘ΎΉ‘»Μ…±…Υ(NK)œΗΑϊ÷–±μ¥οΥ°ΤΫΖ«≥ΘΒΆ, ‘Ύ―Σ–ΓΑεΚΆΚλœΗΑϊ…œ≤Μ¥φ‘Ύ. Φχ”ΎCD37 ―Γ‘ώ–‘±μ¥ο”Ύ B œΗΑϊ, CD37 ≥…ΈΣ÷ΈΝΤ B œΗΑϊΕώ–‘÷ΉΝω(CLL)ΒΡΝΦΚΟΑ–±ξ. Κσά¥”–»ΥΖΔœ÷, CD37 “≤Ω…‘Ύ TœΗΑϊΝήΑΆΝωΓΔAML ΚΆ±μ¥ο CD34ΘΪCD38Θ≠ΒΡ AML Η…œΗΑϊ÷–±μ¥ο, ΒΪ‘Ύ’ΐ≥ΘΒΡ±μ¥οΒΡ CD34ΘΪCD38Θ≠Η…œΗΑϊΚή…ΌΜρΟΜ”–±μ¥ο. AGS-67E ΒΡΉς”ΟΜζ÷Τ”κ Brentuxmab VedotinœύΥΤ, Ήœ»Ά®ΙΐΩΙΧε Ε±πΑ–Βψ, –Έ≥…Η¥ΚœΈοΫχ»κœΗΑϊΡΎ, ‘Ό‘Ύ»ήΟΗΧε÷–±ΜΒΑΑΉΟΗΥ°Ϋβ, ΆΖ≈ MMAE, Ι MMAE‘ΎœΗΑϊΡΎΖΔΜ”ΕΨ–‘Ής”Ο. ΧεΡΎΆβ ‘―ι±μΟς, AGS-67E Ω…“‘“÷÷Τ AML œΗΑϊœΒ. “‘…œΫαΙϊΨυ±μΟς AGS-67E “‘CD37 ΈΣΑ–±ξ÷ΈΝΤ AMLΓΔΝήΑΆœΗΑϊΑΉ―Σ≤ΓΒ»―Σ“ΚΕώ–‘÷ΉΝω«ΑΨΑΙβΟς[38].
ΈΣΤάΦέ‘Ύ”–/Έό…ζ≥Λ“ρΉ”(GF)‘ΛΖά«ιΩωœ¬, AGS-67E÷ΈΝΤΗ¥ΖΔ–‘/Ρ―÷Έ–‘Ζ«ΜτΤφΫπΝήΑΆΝωΜΦ’ΏΒΡΑ≤»Ϊ–‘ΓΔ”––ß–‘ΦΑ“©¥ζΕ·ΝΠ―ß(PK), Ϋχ––ΝΥΦΝΝΩΒί‘ωΓΔΕύ÷––ΡΓΔΔώΤΎΝΌ¥≤ ‘―ι[39]. 13ΟϊΜΦ’ΏΖ÷ΈΣ7ΗωΦΝΝΩΉι(ΈόGF: 0.05ΓΪ1.2 mg/kg, ”– GF: 1.2 mg/kg)Ϋχ––ΒΞ“©÷ΈΝΤ, 3 ÷ή/¥Έ. ΟΜ”–GF ‘ΛΖάΜΦ’ΏΒΡΉν¥σΡΆ ήΦΝΝΩ(MTD)≥§Ιΐ 1.2 mg/kg, ‘ΎΒΎ“Μ¥ΈΗχ“©ΚσΒΡ 8ΓΪ15 d Ιέ≤λΒΫ 3 ΟϊΜΦ’Ώ≥ωœ÷―œ÷Ί÷––‘ΝΘœΗΑϊΦθ…Ό. ‘ΎΗχ”η 1.2 mg/kg ΦΝΝΩΉι÷–“ΜΟϊΜΦ”–Ο÷¬ΰ–‘¥σ B œΗΑϊΝήΑΆΝωΒΡΜΦ’ΏΆξ»ΪΜΚΫβ. ‘Ύ 1.2 mg/kg ±, S-67E ΚΆ”Έάκ MMAE ΒΡΑκΥΞΤΎΖ÷±πΈΣ 1.44ΓΪ3.08 ΚΆ2.34ΓΪ3.64 d. ΤδΫαΙϊ±μΟς 3 ÷ή/Ηχ“© AGS-67E ΨΏ”–÷ΈΝΤΝήΑΆΝωΒΡΜν–‘«“Α≤»Ϊ–‘ΝΦΚΟ.
1.3 Solidotin (TZT-1027)
Solidotin « Dolastatin 10 ΒΡΚœ≥…―ή…ζΈο, Ήν≥θ”…Teikoku Hormone ΩΣΖΔ÷ΈΝΤΆμΤΎΜρΉΣ“ΤΒΡ»μΉι÷·»βΝω(STS)ΜρΖ«–ΓœΗΑϊΖΈΑ©(NSCLC), ¥Π”ΎΝΌ¥≤ II ΤΎ―–ΨΩΫΉΕΈ. Solidotin Ά®Ιΐ”κΈΔΙήΒΑΑΉΫαΚœ, “÷÷ΤΈΔΙήΒΑΑΉΒΡΨέΚœ, ΙœΗΑϊ G2-M ΤΎΖ÷Μ·ΆΘ÷Ά, ΒΦ÷¬œΗΑϊΒρΆω[40]. ≤Δ«“, Solidotin Ω…“‘ΙΞΜςΆμΤΎ÷ΉΝωΝΦΚΟΒΡ―ΣΙήœΒΆ≥, Ήη÷Ι―ΣΙή…ζ≥…, …±Υά÷ΉΝωœΗΑϊ[41]. Watanabe Β»[42]ΖΔœ÷ Solidotin ΒΡΩΙ÷ΉΝωΜν–‘“Σ”≈”Ύ≥Λ¥Κ–¬ΦνΓΔ5-ΖζΡρύΉύΛΚΆΥ≥¬»Α±≤§. ‘ΎΫαΙΙ…œ, Solidotin «”Ο±Ϋ““ΑΖ»Γ¥ζDolastatin 10ΒΡDoeΤ§ΕΈΕχΒΟΒΫΒΡ, ΥϋΒΡΚœ≥… «“‘”κ Dil Τ§ΕΈœύΙΊΝΣΒΡΜ·ΚœΈο(24)Κœ≥… Dov-Val-Dil-Dap-OBzl (32), »ΐκΡ 32 Ϋχ––Ά―±ΘΜΛ‘Ό”κ±Ϋ““ΑΖ(33)Ζ¥”ΠΒΟΒΫ Solidotin (Scheme 7)[15].
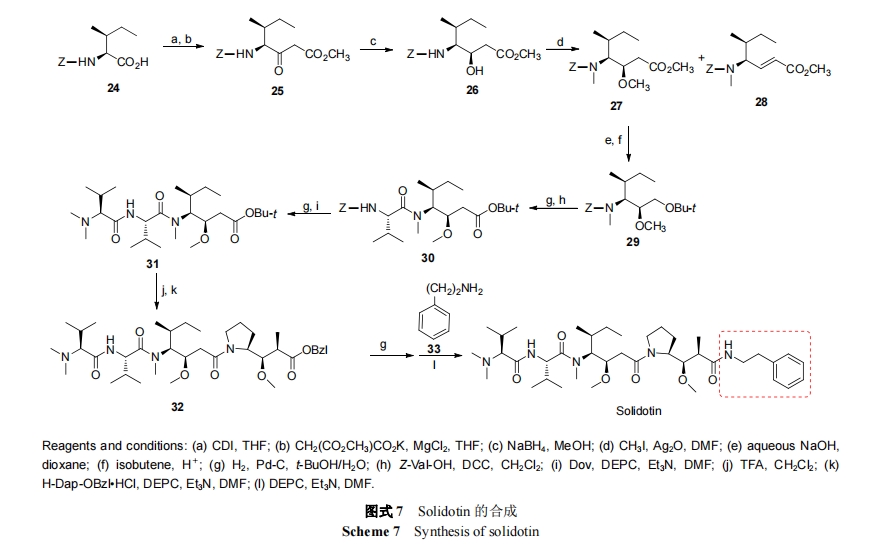
ΈΣΤάΦέ Solidotin ÷ΈΝΤΆμΤΎΜρΉΣ“Τ»μΉι÷·»βΝωΒΡΑ≤»Ϊ–‘ΦΑ”––ß–‘, Patel Β»[43]Ρ…»κ 28 ΟϊΖϊΚœΜΦ’ΏΫχ–– II ΤΎΝΌ¥≤ ‘―ι. Solidotin Ήι, ÷ΉΝωΫχ’ΙΒΡ÷–÷Β ±ΦδΈΣ 44 d (95%÷Ο–≈«χΦδ[95% CI], 43.0ΓΪ54.0), ÷–ΈΜ…ζ¥φΤΎΈΣ 178 d (95% CI, 134.0ΓΪ317.0). ≥ΘΦϊΒΡ≤ΜΝΦΖ¥”Π «÷––‘ΝΘœΗΑϊΦθ…ΌΓΔΤΘάΆΚΆ±ψΟΊ. ±Ψ ‘―ιΫαΙϊ±μΟς, Solidotin Α≤»Ϊ–‘ΦΑΡΆ ή–‘ΝΦΚΟ, ΒΪ≤Δ≤ΜΡή”––ßΜΚΫβ÷ΉΝωΫχ’Ι. Νμ“Μœν”…Riely Β»[44]Ήι÷·ΒΡ II ΤΎΝΌ¥≤ ‘―ι―Γ‘ώΝΥ 32 ΟϊΖϊΚœΧθΦΰΒΡΜΦ’Ώ. “‘ 28 d ΈΣ“ΜΗχ“©÷ήΤΎ, ‘ΎΒΎ 1 d ΚΆΒΎ 8 d Ηχ”ηΜΦ’Ώ2.4 mg/m2 Solidotin. ‘Ύ ‘―ι÷–, Ήν≥ΘΦϊΒΡ―œ÷Ί≤ΜΝΦΖ¥”Π «ΑΉœΗΑϊΦθ…ΌΚΆ÷––‘ΝΘœΗΑϊΦθ…Ό. 4 ΟϊΜΦ’Ώ‘ΎΫ” ήSolidotin ΒΡ 30 d ΡΎΥάΆω, 3 ΟϊΜΦ’Ώ≤Γ«ιΫχ––, 1 ΟϊΜΦ’Ώ≥ωœ÷ΖΈ―ΉΚΆ÷––‘ΝΘœΗΑϊΦθ…Ό. ‘―ιΫαΙϊ±μΟς, “‘ 28 d ΈΣ“Μ÷ήΤΎ, ‘ΎΒΎ1 dΚΆΒΎ8 dΗχ”ηΜΦ’Ώ2.4 mg/m2 Solidotin, ΜΦ’Ώ≤Γ«ιΈ¥≥ωœ÷ΩΆΙέΜΚΫβ, Έ¥¥οΒΫ‘ΛΤΎ÷ΈΝΤ–ßΙϊ.
1.4 Dolastatin 15 ―ή…ζΈο
Dolastatin 15(ΆΦ 5)Ω…“‘”––ß“÷÷Τ÷ΉΝω…ζ≥Λ, ΒΪ”…”ΎΤδΫαΙΙΗ¥‘”ΓΔΜ·―ßΚœ≥…≤ζ¬ ΒΆΦΑΥ°»ή–‘≤νΒ»‘≠“ρœό÷ΤΝΥDolastatin 15 ΒΡΩΆΙέΝΌ¥≤ΤάΦέ. ΈΣΝΥΫβΨω Dolastatin 15ΒΡ’β–©±ΉΕΥ, Κœ≥…ΝΥ Dolastatin 15 ΒΡ―ή…ζΈο[45]. Τδ÷–, Tasidotin ΚΆ Cemadotin “―Ϋχ»κΝΌ¥≤ ‘―ι.
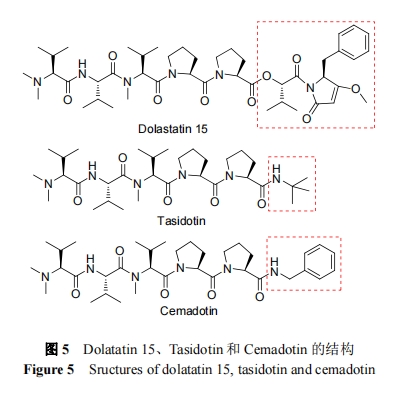
1.4.1 Tasidotin, Synthadotin (IXL-651)
Tasidotin « Dolastatin 15 ΒΡΒΎ»ΐ¥ζΚœ≥…―ή…ζΈο. ‘ΎΫαΙΙ…œ, Tasidotin ‘Ύτ»ΜυΡ©ΕΥ”Ο εΕΓΜυΑΖ»Γ¥ζ Dolastatin 15ΒΡθΞΜυΆ≈(ΆΦ5). TasidotinΒΡ÷ς“ΣΉς”ΟΜζ÷Τ «“÷÷ΤΖΡ¥ΗΧεΒΡ–Έ≥…. Tasidotin ‘ΎΧεΡΎΒΡ÷ς“Σ¥ζ–Μ≤ζΈο « N,N- ΕΰΦΉΜυγ”Α±Υα-γ”Α±Υα-N-ΦΉΜυγ”Α±Υα-Η§Α±θΘΜυ-Η§Α±Υα(P5), P5 “÷÷ΤΈΔΙήΒΑΑΉΨέΚœΒΡΜν–‘“Σ±» Tasidotin ΗΏ, ΒΪΉςΈΣœΗΑϊΕΨ–‘ΦΝΒΡΜν–‘“Σ±» Tasidotin ΒΆ[46].
ΈΣΤάΦέ Tasidotin ΒΡΑ≤»Ϊ–‘ΓΔΡΆ ή–‘ΚΆ“©¥ζΕ·ΝΠ―ß, Mita Β»[47]Ρ…»κ 30 Οϊ ή ‘’ΏΫχ–– I ΤΎΝΌ¥≤ ‘―ι. ‘Ύ 7.8 ÷Ν62.2 mg/m2ΒΡΦΝΝΩΖΕΈß…η÷Ο 6ΗωΦΝΝΩΉι, 1¥Έ/÷ήΗχ“©, ≥÷–χ 82 ΗωΝΤ≥Χ. ‘ΎΥυ”–ΦΝΝΩΥ°ΤΫ…œ, ΖΔ…ζ“ΜΑψ ADR ”–ΗΙ–ΚΚΆ≈ΜΆ¬, Ζ«―Σ“Κ―ßΕΨ–‘Ά®≥ΘΈΣ«α÷Ν÷–Ε»«““Ή”ΎΩΊ÷Τ. Tasidotin “©¥ζΕ·ΝΠ―ß ««αΕ»Ζ«œΏ–‘ΒΡ, Εχ¥ζ–ΜΕ·ΝΠ―ß «œΏ–‘ΒΡ. Ζ«–ΓœΗΑϊΖΈΑ©ΜΦ’ΏΖΰ“©Κσ≤Γ«ι«αΈΔΜΚΫβ, ΗΈœΗΑϊΑ©ΜΦ’Ώ‘Ύ 11 Ηω‘¬ΡΎ≤Γ«ιΈ»Ε®.
1.4.2 Cemadotin (LU-103793)
Cemadotin « Dolastatin 15 ΒΡΥ°»ή–‘Κœ≥…―ή…ζΈο. ”… Abbott ΙΪΥΨ―–ΖΔ, ”Ο”Ύ÷ΈΝΤΑ©÷Δ, ‘χ¥Π”ΎΝΌ¥≤ II ΤΎ, œ÷“―±Μ÷’÷Ι. ‘ΎΫαΙΙ…œ, Cemadotin‘Ύ CΕΥ”Ο±ΫΦΉΑΖ»Γ¥ζΝΥ Dolastatin 15 ΒΡθΞΜυΆ≈(ΆΦ 5)[45]. Cemadotin ΒΡΫαΚœΈΜΒψ«χ±π”Ύ≥Λ¥ΚΦν, ΤδΫχ»κœΗΑϊΚσ, “÷÷ΤΈΔΙήΒΑΑΉΒΡΨέΚœ, ΙœΗΑϊ÷ήΤΎΆΘ÷Ά‘Ύ G2-M ΫΉΕΈ[48].
“Μœν II ΤΎΝΌ¥≤ ‘―ιΤάΙάΝΥ Cemadotin ‘ΎΉΣ“Τ–‘»ιœΌΑ©ΜΦ’Ώ÷–ΒΡΑ≤»Ϊ–‘ΚΆ”––ß–‘. ‘ΎΩ…ΤάΦέΒΡ 23 ΟϊΜΦ’Ώ÷–, 11 ΈΜΜΦ’ΏΖΔ…ζΝΥ―œ÷Ί÷––‘ΝΘœΗΑϊΦθ…Ό÷Δ, ΤδΥϋ“ΜΑψADR ”–ΖΠΝΠΓΔΩΎ«Μ―ΉΓΔΦΓΆ¥ΦΑ―Σ«εΒ®ΚλΥΊ…ΐΗΏ, ÷ς“ΣADR ΈΣΗΏ―Σ―Ι. ‘Ύ ‘―ιΤΎΦδ, ΜΦ’Ώ≤Γ«ιΈόΩΆΙέΜΚΫβ, Ι ÷’÷Ι ‘―ι[49].
2 ”σ¬ίΕΨΥΊ
ΉΕ–ΈΈœ≈Θ÷ς“Σ…ζΜν‘Ύ»»¥χΚΘ”ρ, ΥϋΟ«Ά®Ιΐ–Έ≥…ΕΨ“Κά¥±ΘΜΛΉ‘…μΦΑΝ‘ ≥. ‘ΎΕΨ“Κ÷–ΑϋΚ§÷¬ΥάΒΡΕΨΥΊΓΣΓΣ”σ¬ίΕΨΥΊ. ”σ¬ίΕΨΥΊ «“Μ÷÷–ΓΖ÷Ή”, ΫαΙΙΈ»Ε®, ΨΏ”–ΗΏΕ»Α–œρΧΊ“λ–‘ΓΔΗΜΚ§”–ΕΰΝρΦϋΒΡΜν–‘ΕύκΡ, Ά®≥Θ”… 12 ΒΫ 41ΗωΑ±ΜυΥαΙΙ≥…, «““Ή”ΎΚœ≥…[50]. Υϋ÷ς“ΣΑ–œρ”ΎΡΛΒΑΑΉ, ΧΊ±π «Βγ―ΙΜρ≈δΧεΟ≈ΩΊάκΉ”Ά®Βά, ΉΣ‘ΥΒΑΑΉΜρ G ΒΑΑΉ≈ΦΝΣ ήΧε. ΥϋΩ…“‘”Ο”Ύ÷ΈΝΤ…ώΨ≠–‘ΧέΆ¥ΓΔΑ©÷ΔΆμΤΎΒΡ¬ΐ–‘ΧέΆ¥“‘ΦΑΤδΥϊΧέΆ¥. ”…”ΎΥϋΨΏ”–ΫœΗΏΒΡ…ζΈοΜν–‘ΦΑΑ–œρΧΊ“λ–‘«““Ή”ΎΚœ≥…, ΙΒΟ”σ¬ίΕΨΥΊ”–Άϊ≥…ΈΣœ»ΒΦΜ·ΚœΈοΜρ“©Έο[51]. 21 άΦΆ≥θΤΎ, Έ“ΙζΤί’ΐΈδ‘Κ ΩΩΈΧβΉιΩΣ ΦΝΥΕ‘Ηςάύ”σ¬ίΕΨΥΊΒΡ―–ΨΩ, Ϋχ––Ζ≠“κΚσ–ό Έ, ΧΫ≤βΤδΙΠΡή, ΈΣΩΣΖΔΗΏ–ßΕχΧΊ“λΒΡ…ώΨ≠“©Έο¥ρœ¬Μυ¥Γ. ΡΩ«Α, ΠΊ-”σ¬ίΕΨΥΊΒΡΚœ≥…–Έ ΫΓΣΓΣΤκΩΦ≈ΒκΡ(Ziconotide, Scheme 8)“―…œ –.
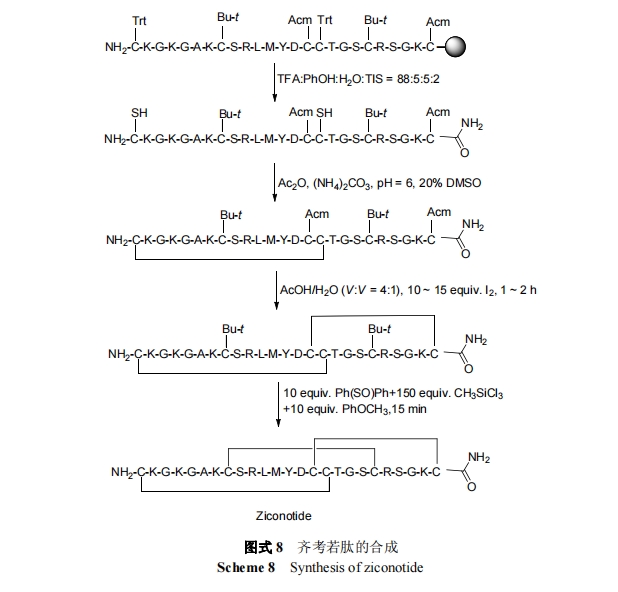
ΤκΩΦ≈ΒκΡ(“‘«Α≥Τ SNX-111) «ά¥‘¥”ΎΧΪΤΫ―σΒΡΜΟ”σ¬ίΕΨ“Κ÷–ΨΏ”–«ΉΥ°–‘ ΠΊ-MVIIA ”σ¬ίΕΨΥΊΒΡΚœ≥…–Έ Ϋ. Υϋ”Ύ 2004 Ρξ 12 ‘¬Ψ≠ FDA ≈ζΉΦ…œ –, ”Ο”Ύ≥ΘΙφ÷ΈΝΤ≤ΜΡΆ ήΜρΡ―”Ύ÷ΈΝΤΒΡ¬ΐ–‘―œ÷Ί–‘Ν§–χ« ΡΎ≤Γ±δΜΦ’ΏΒΡΧέ Ά¥[52]. “ΜœνΉ®άϊ[53]±®ΒάΝΥΤδΚœ≥…¬ΖœΏ(Scheme 8), “‘Fmoc-Α±Μυ ς÷§ΈΣΙΧœύ‘ΊΧε, ¥” C ΕΥΒΫ N ΕΥ“ά¥Έ”ΟΥθΚœΖ¥”ΠΝ§Ϋ” 25 Ηω≤ύΝ¥±ΘΜΛΒΡΑ±ΜυΥα, –Έ≥…ΕΰΝρΦϋΒΡ»ΐΉιCys Ζ÷±πΝ§Ϋ” Trt, Acm ΚΆ t-Bu ±ΘΜΛΜυ, ΫΪ ς÷§Ϋχ––œΏ–‘«–Ην, Ά§ ±Ά―»Ξ≥ΐ Cys (Acm)ΚΆ Cys (t-Bu)ΆβΒΡΥυ”–Α±ΜυΥαΒΡ≤ύΝ¥±ΘΜΛΜυ, ―θΜ·œΏ–‘κΡ–Έ≥…ΒΎ“ΜΕ‘ΕΰΝρΦϋ, ΒΟΒΫΒΞΕΰΝρΜΖκΡ, »ΜΚσ“ά¥ΈΆ―≥ΐ Cys (Acm)ΚΆ Cys (t-Bu)÷–ΒΡAcm ΚΆ t-Bu, ‘ΎΆ―≥ΐ ±Ά§ ±ΜΖΜ·, ΒΟΒΫ»ΐΕΰΝρΜΖκΡ, ΉνΚσΨ≠ HPLC ¥ΩΜ·, Ε≥Η…ΒΟΒΫΤκΩΦ≈ΒκΡ.
ΤκΩΦ≈ΒκΡΑϋΚ§25ΗωΑ±ΜυΥα, ΗΜΚ§”–»ΐΗωΕΰΝρΦϋ, Τδ“©άμΜν–‘“άάΒ”Ύ’β–©Άξ’ϊΒΡΕΰΝρΦϋ, ’β–©ΕΰΝρΦϋ“≤ «Ζ÷Ή”»ΐΈ§ΫαΙΙΒΡΨωΕ®“ρΥΊ. 3 Ι… Π¬ ’έΒΰΩ…Ϋχ“Μ≤ΫΈ»Ε®Ω’ΦδΫαΙΙ, ‘ω«ΩκΡΕ‘ΟΗΒΡΩΙ–‘, “≤Ω…Ρή‘ω«ΩΫαΚœΝΠΚΆ–ßΝΠ. ΒΑΑΉ±μΟφ”κ¥χΒγΚΆΦΪ–‘Α±ΜυΥα≤ύΝ¥Ν§Ϋ”, ΙΒΟΤκΩΦ≈ΒκΡΗΏΕ»«ΉΥ°. ΤκΩΦ≈ΒκΡΒΡΫαΙΙΑΒ ΨΤδΩ…“‘”Ο”Ύ≥…“©[54].ΤκΩΦ≈ΒκΡ «“Μ÷÷–ßΙϊΖ«≥ΘΚΟΒΡΖ«ΑΔΤ§άύ’ρΆ¥“©, ΥϋΕ‘ N–ΆΒγ―ΙΟτΗ–ΗΤΆ®ΒάΨΏ”–ΫœΗΏΒΡ«ΉΚΆΝΠ, Ω…“‘”––ßΒΊΉηΕœΗΤάκΉ”ΒΡΝςΆ®. “ρ¥Υ, ΥϋΩ…“‘Ά®Ιΐ”––ßΚΆ―Γ‘ώ–‘ΉηΕœ N–‘ΗΤάκΉ”Ά®Βά, ¥”ΕχΩΊ÷ΤΕύ÷÷ΆΜ¥Ξ ΆΖ≈ΒΡ…ώΨ≠Βί÷ . ΥϋΩ…Ρή‘ΎΦΙΥηΥ°ΤΫ…œ÷–ΕœΝΥΧέΆ¥–≈Κ≈ΒΡ¥ΪΒί[55].
ΈΣΤάΙά« ΡΎΤκΩΦ≈ΒκΡΕ‘≥ΘΙφ÷ΈΝΤΡ―÷ΈΒΡΧέΆ¥ΜΦ’ΏΒΡΑ≤»Ϊ–‘ΚΆ”––ß–‘Ϋχ––ΝΥΥΪΟΛΓΔΑ≤ΈΩΦΝΥφΜζΕ‘’’ ‘ ―ι[56], ‘―ι―Γ‘ώ 111 άΐΑ©÷ΔΜρ AIDS ΜΦ’Ώ, “‘ 2ΓΟ1 ΒΡ±»άΐΥφΜζΖ÷≈δ÷ΝΤκΩΦ≈ΒκΡΉιΚΆΑ≤ΈΩΦΝΉι. ‘ΎΩ…ΤάΦέΒΡΜΦ’Ώ÷–, ΤκΩΦ≈ΒκΡΉι÷– 52.9%ΒΡΜΦ’ΏΧέΆ¥ΜΚΫβ, ‘ΎΑ≤ΈΩΦΝΉι÷–, ÷Μ”– 17.5% (PΘΦ0.001), ≤Δ«“‘ΎΫ” ήΤκΩΦ≈ΒκΡΒΡΜΦ’Ώ÷–, ”– 5 ΈΜΜΦ’Ώ¥οΒΫΝΥΆξ»ΪΜΚΫβ. ΫαΙϊ±μΟςΤκΩΦ»τκΡΕ‘”ΎΜΚΫβΡ―÷Έ–ΆΧέΆ¥ΨΏ”–ΚήΚΟΒΡΝΤ–ß.
3 ΚΘ« ΥΊ
1981 Ρξ Rinehart Β»¥”ΡΛΚΘ« ÷–Χα»Γ≥ω“ΜœΒΝ–ΜΖ–ΈΥθκΡάύΜν–‘Έο÷ , ΟϋΟϊΈΣ Didemnins A, B, C, ΥϋΟ«ΨΏ”–ΫœΗΏΒΡΩΙ÷ΉΝωΜν–‘. Τδ÷–Μν–‘ΉνΗΏΒΡ « Didemnin B, Υϋ≥…ΈΣΟάΙζΒΎ“ΜΗωΫχ»κΝΌ¥≤ ‘―ιΒΡ‘¥Ή‘ΚΘ―σΒΡΩΙ÷ΉΝωΧλ»Μ≤ζΈο. ΒΎΕΰ¥ζ DidemninΓΣΓΣΆ―«β Didemnin B (Plitidepsin)“≤’ΐ‘ΎΫχ––ΝΌ¥≤ ‘―ι[57]. Plitidepsin ”κ Didemnin B ‘ΎΖ÷Ή”ΫαΙΙ…œΫω≤νΝΫΗω«β(ΆΦ 6).
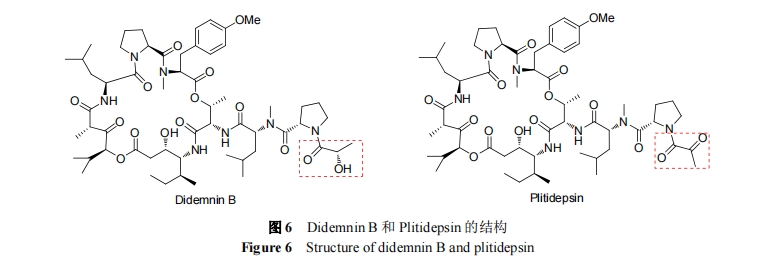
3.1 Didemnin B
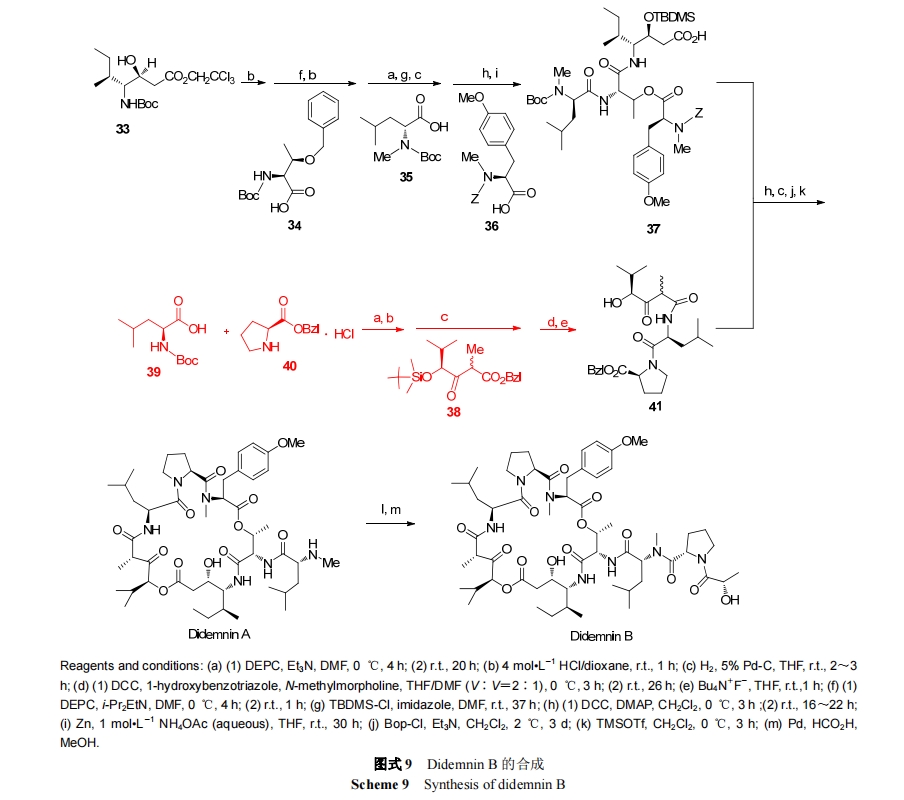
―–ΨΩ»Υ‘±Ε‘ Didemnin B ÷ΈΝΤ«ΑΝ–œΌΑ©ΓΔΖ«–ΓœΗΑϊΖΈΑ©ΓΔΙ«ΥηΝωΚΆΚΎΥΊΝωΒ»Εύ÷÷÷ΉΝωΫχ––ΝΥ I ΤΎΓΔII ΤΎΝΌ¥≤ ‘―ι. ΤδΉς”ΟΜζ÷Τœ÷‘ΎΜΙΈ¥Ος»Ζ≤ϊ ω, ΒΪΥϋΥΤΚθ”κ“÷÷ΤΒΑΑΉ÷ Κœ≥…”–ΙΊ, ‘ΎΫœ–Γ≥ΧΕ»…œΩ…Ρή”κ“÷÷Τ DNA ΚΆRNA Κœ≥…”–ΙΊ[58]. Didemnin B ΨΏ”––¬–ΆΒΡΜΖΉ¥ΫαΙΙ, Αϋά®“ΜΗω“÷ΈΗΟΗΑ±Υα, ‘ωΦ”ΝΥ Didemnin B ΒΡΈ»Ε®–‘[59]. Hamada Β»[60]±®ΒάΤδΚœ≥…¬ΖœΏ(Scheme 9), Ά®Ιΐ(2RS,4S)- Hip-(S)-Leu-(S)-Pro-OBzl (42) ΚΆ Boc-(R)-MeLeu-(S)- Thr[Z-(S)-MeTyr(Me)]-(3S,4R,5S)-Ist(TBDMS)-OH (38)≈ΦΝΣ≤ΔΜΖΜ·, »ΜΚσ»Ξ±ΘΜΛ, ΒΟΒΫ Didemnin A, »ΜΚσΫΪΤδΉΣΜ·ΈΣ Didemnin B.
ΈΣΤάΦέ Didemnin B ÷ΈΝΤΗ¥ΖΔΜρΖΔ’ΙΒΡ NHL ΜΦ’ΏΒΡΙΠ–ßΚΆΕΨ–‘, Ϋχ––ΝΥΝΌ¥≤IIΤΎ ‘―ι. ‘―ι―Γ‘ώΝΥ51ΟϊΜΦ’Ώ. ‘Ύ ‘―ι÷–, ΜΦ’ΏΕΦ≤ζ…ζΝΥ 3ΓΪ4 ΦΕΒΡ≤ΜΝΦΖ¥”Π, ΕΨ–‘Ϋœ¥σ, “ρ¥ΥΆΘ÷ΙΝΥ Β―ι[61].
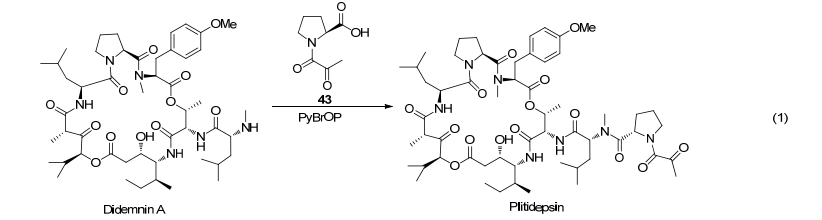
3.2 Plitidepsin (Aplidin)
Plitidepsin “ύ≥Τ Aplidin, ”… PharmaMar ―–ΖΔ. Υϋ «¥”ΒΊ÷–ΚΘΚΘ« Aplidium albicans ÷–Ζ÷άκΒΟΒΫΒΡΩΙ÷ΉΝωΜν–‘Χλ»Μ≤ζΈοΒΡΚœ≥…–Έ Ϋ. ΥϋΨ≠ FDA ΚΆ≈Ζ÷όΈ·‘±Μα≈ζΉΦΈΣ÷ΈΝΤΕύΖΔ–‘Ι«ΥηΝωΒΡΙ¬Ευ“©. ‘ΎΜν–‘≈®Ε» ±, Plitidepsin œ‘ Ψ±» Didemnin B ΗϋΒΆΒΡΕΨ–‘[62]. Plitidepsin ΨΏ”–ΗϋΗΏΒΡ÷ΈΝΤΦέ÷Β, œ÷¥Π”Ύ÷ΈΝΤΕύΖΔ–‘Ι«ΥηΝωΒΡΝΌ¥≤ΔσΤΎΫΉΕΈ.
Plitidepsin Ής”ΟΜζ÷Τ «Ά®Ιΐ”κœΗΑϊΡΛ±μΟφΗΏ«ΉΚΆΝΠΒΡΈΜΒψΫαΚœΚσ, ΦΛΜν Rac1, P38/MAPK ΚΆ JNK –≈Κ≈¥ΪΒΦ, Fas/CD95 ΉΣ“Τ÷Ν÷§ΖΛ¥Π, Ήν÷’“ΐΤπΑκκΉΑ±ΥαΒΑΑΉΟΗΒΡΦΛΜν, ΙœΗΑϊΒρΆω[63]. Plitidepsin Ά®Ιΐ(S)-Pro-Pyr (43)”κDidemnin A ÷–ΒΡ(R)-N-ΦΉΜυ-Leu ≤ύΝ¥÷–ΒΡΑ±ΜυΫαΚœ–Έ≥…(Eq. 1)[64].
“ΜœνΕύ÷––ΡΓΔΒΞΉιΓΔΩΣΖ≈«“ΨΏ”–«Α’Α–‘ΒΡ II ΤΎΝΌ¥≤ ‘―ι[65]ΤάΙάΝΥ plitidepsin ÷ΈΝΤΗ¥ΖΔ/Ρ―÷Έ–‘ΕύΖΔ–‘Ι«ΥηΝω(MM)ΜΦ’ΏΒΡΑ≤»Ϊ–‘ΚΆ”––ß–‘. ‘ΎΩ…ΤάΦέΒΡ 47 ΟϊΜΦ’Ώ÷–, ΒΞΕά Ι”Ο plitidepsin ±ΒΡΉήΜΚΫβ¬ (OR)ΈΣ 13%, ”κΒΊ»ϊΟΉΥ…ΝΣΚœ Ι”Ο ±ΈΣ 22%. “ΜΑψ ADR ”–ΤΕ―Σ(29%)ΓΔ―Σ–ΓΑεΦθ…Ό÷Δ(18%)ΓΔΤΘάΆ(16%)ΓΔΦΓ»βΕΨ–‘(6%)ΚΆΥ≤ ±±ϊΑ±ΥαΑ±ΜυΉΣ“ΤΟΗ/ΧλΕ§Α±ΥαΑ±ΜυΉΣ“ΤΟΗ(27%)ΚΆΦΓΥαΝΉΥαΦΛΟΗ(23%)‘ωΦ”. ’β±μΟςΒΞ”ΟΜρ”κΒΊ»ϊΟΉΥ…ΝΣ”Οplitidepsin ÷ΈΝΤΗ¥ΖΔ/Ρ―÷Έ–‘ΕύΖΔ–‘Ι«ΥηΝωΜΦ’Ώ «Α≤»ΪΓΔ”––ßΒΡ.
4 ΚΘΟύΜν–‘κΡ
Hemiasterlin(ΆΦ 7)Ήν≥θ «¥”ΡœΖ«ΚΘΟύ Hemiasterella minor ÷–Χα»Γ≥ωΒΡ»ΐκΡ[66]. ΤδΉς”ΟΜζ÷Τ «Ά®Ιΐ”κΈΔΙήΒΑΑΉΫαΚœ, ΙΈΔΙήΫβΨέ, ”ΑœλΖΡ¥ΗΧεΒΡ–Έ≥…Εχ≤ζ…ζœΗΑϊΕΨ–‘. Υϋ‘ΎΧεΆβΕ‘–Γ σΑΉ―Σ≤ΓœΗΑϊ P388 (ED50ΘΫ4.57ΓΝ10Θ≠5µg/mL)ΓΔ»Υ»ιœΌΑ©œΗΑϊ MCF-7 (ED50ΘΫ0.089 µg/mL)ΓΔ»Υ≥…ΫΚ÷ ΝωœΗΑϊ/–«–ΈΫΚ÷ ΝωœΗΑϊ U373 (ED50ΘΫ0.012µg/mL)ΓΔ»Υ¬―≥≤Α©œΗΑϊ HEY(ED50ΘΫ0.0014 µg/mL)ΚΆΧεΡΎΕ‘–Γ σΒΡ P388 œΗΑϊΨΏ”–Μν–‘[67]. ΥϋΝΦΚΟΒΡΜν–‘ ΙΒΟΥϋ”–Άϊ≥…ΈΣœ»ΒΦΜ·ΚœΈο. »ΥΟ«ΈΣΝΥΧΫ≤βΥϋΒΡΫαΙΙ”κΜν–‘ΒΡΙΊœΒ, Κœ≥…ΝΥ“ΜœΒΝ–―ή…ζΈο, Τδ÷–HTI-286ΒΡΜν–‘±»Χλ»Μ≤ζΈο Hemiasterlin Μν–‘ΚΟ, œ÷“―Ψ≠Ϋχ»κΝΌ¥≤ ‘―ιΫΉΕΈ[68].
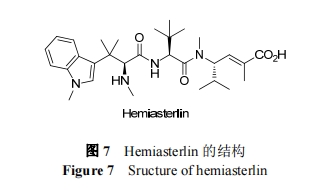
4.1 HTI-286
HTI-286 ”…Μ‘»πΙΪΥΨ―–ΖΔ, ”Ο”Ύ÷ΈΝΤΑ©÷Δ. Niu Β»[69]±®ΒάΝΥΤδΚœ≥…¬ΖœΏ(Scheme 10), ΥϋΩ…“‘ΦΉΜυΜ·ΒΡ±ϊΆΣΥα―ή…ζΈο44ΉςΈΣ‘≠Νœ, ‘Ύ«βΜ·ΡΤ¥φ‘Ύœ¬, ”κΒβΦΉΆιΖ¥”ΠΒΟΒΫΌ…ΕΰΦΉΜυΜ·ΚœΈο 45, 45 Ϋχ––ΜΙ‘≠Α±Μ·ΒΟΒΫΤ§ΕΈ 46, 46 ”κΕΰκΡ 47 ≈ΦΝΣΒΟΒΫΖ«Ε‘”Π“λΙΙΧε 48, ΫΪΤδΆ®ΙΐHPLCΖ÷άκΒΟΒΫ(S,S,S)-48, ‘ΎLiOH¥φ‘Ύœ¬, ΫΪ(S,S,S)-48Ϋχ––Υ°ΫβΦ¥Ω…ΒΟΒΫ HTI-286. ‘Ύ―–ΨΩ HTI-286 ΫαΙΙ”κΜν–‘ΒΡΙΊœΒ ±ΖΔœ÷, HTI-286 ΫαΙΙ÷–ΉνΉσ≤ύΑ±ΜυΥα÷–ΒΡΦν–‘ΜυΆ≈ «ΙΊΦϋΜυΆ≈, ’β“ΜΖΔœ÷ΈΣ–¬–Ά―ή…ζΈοΒΡΚœ≥…¥ρœ¬Μυ¥Γ[70]. HTI-286 Ά®Ιΐ“÷÷ΤΈΔΙήΒΑΑΉΒΡΨέΚœ, ΤΤΜΒœΗΑϊ÷–ΒΡΈΔΙήΉι÷·, ≤Δ”’ΒΦ”–ΥΩΖ÷Ν―ΆΘ÷Ά, ΙœΗΑϊΒρΆω, ≤Δ«“Υϋ≤Δ≤Μ”κΕύ“©ΡΆ“©ΒΑΑΉ P-Χ«ΒΑΑΉΫαΚœ, ±»Ήœ…Φ¥ΦΓΔΕύΈςΥϊ»ϋΓΔ≥Λ¥ΚΦνΒ»ΩΙ÷ΉΝω“©Έο–ßΙϊΚΟ[71]. ΝΌ¥≤«Α―–ΨΩ±μΟς, ‘ΎΧεΆβΚΆΧεΡΎ HTI-286 Ε‘”Ύ÷ΈΝΤΗΈΑ©ΓΔ«ΑΝ–œΌΑ©ΚΆΑρκΉΑ© «”––ßΒΡ[72]. ’β“≤±μΟς HTI-286 ΨΏ”–ΉςΈΣ÷ΈΝΤΕύ÷÷Εώ–‘÷ΉΝω“©ΈοΒΡ«±ΝΠ.
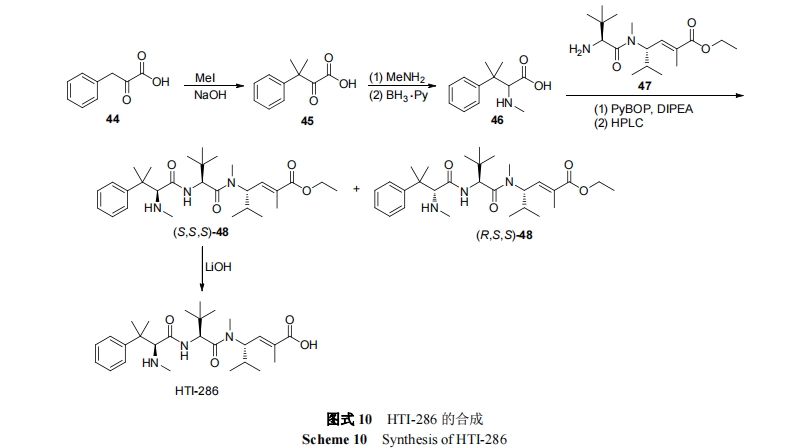
4.2 E7974
E7974 Ά§―υ « Hemiasterlin ΒΡ―ή…ζΈο, ”…Έά≤ΡΙΪΥΨ―–ΖΔ, ”Ο”Ύ÷ΈΝΤ ΒΧεΝω. ‘χ¥Π”ΎΝΌ¥≤ I ΤΎ, œ÷“―÷’÷Ι ‘―ι. ΤδΉς”ΟΜζ÷Τ «Ά®Ιΐ”κ ΠΝ-ΈΔΙήΒΑΑΉΫαΚœ, “÷÷ΤΈΔΙήΒΑΑΉΨέΚœ. ‘ΎΧεΆβœΗΑϊ Β―ι÷–, E7974 Ω…“‘”’ΒΦœΗΑϊΆΘ÷Ά‘Ύ G2-M ΤΎ, ≤ΔΑ–œρΫαΚœΖΡ¥ΗΧε, “÷÷ΤΤδ–Έ≥…[73]. “ΜœνΉ®άϊ±®ΒάΝΥΤδΚœ≥…¬ΖœΏ[74], “‘ N-Boc-N-Me-L-Val-OH (49)ΈΣΤπ Φ‘≠Νœ, Ψ≠ΙΐΖ¥”ΠΚœ≥…Μ·ΚœΈο 53, 54 ”κ 55 Ϋχ––≈ΦΝΣ, Ά―±ΘΜΛ―ΈΥαΜ·ΒΟΒΫ 57. 57 ”κ N-“λ±ϊΜυΏΏΩ…Υα(58)Ψ≠Ιΐ«ΉΚΥΦ”≥…-»Γ¥ζΖ¥”Π, Υ°ΫβΦ¥Ω…ΒΟΒΫΡΩ±ξ≤ζΈο(Scheme 11).
ΈΣΤάΦέΝΥ E7974 ÷ΈΝΤΆμΤΎ ΒΧεΝωΒΡΑ≤»Ϊ–‘ΚΆ”––ß–‘, Ϋχ––ΝΥ I ΤΎΝΌ¥≤ ‘―ι. ‘―ι―Γ‘ώΝΥ 28 ΟϊΜΦ’Ώ, Ζ÷ΈΣ5 ΗωΦΝΝΩΉι(0.18, 0.27, 0.36, 0.45 ΚΆ 0.56 mg/m2; MTD ΈΣ0.45 mg/m2), 28 Χλ/¥Έ, Ψ≤¬ωΉΔ…δΗχ“©(2 ÷Ν 5 min). ‘Ύ 17ΟϊΜΦ”–Ρ―÷Έ–‘Ϋα≥ΠΑ©ΜΦ’Ώ÷–, 7 Οϊ(41%)ΜΦ’Ώ≤Γ«ιΒΟ“‘Έ»Ε®. ÷–ΈΜΈόΫχ’Ι…ζ¥φΤΎΈΣ 1.2 Ηω‘¬, ÷–ΈΜΉή…ζ¥φΤΎΈΣ 6.7Ηω‘¬. ≥ωœ÷―Σ“Κ ADR ”– »÷––‘ΑΉ―Σ«ρΦθ…Ό÷ΔΓΔΤΕ―ΣΓΔΑΉ―Σ«ρΦθ…Ό÷Δ; “ΜΑψ ADR ”–ΤΘάΆΓΔΆΚΆΖ÷ΔΓΔΕώ–ΡΓΔ≈ΜΆ¬ΓΔ±ψΟΊ[75].
5 ΚΘ―σ’φΨζΜν–‘κΡ
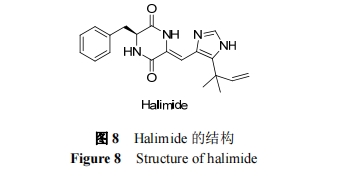
KanohΒ»¥”«ζΟΙ τ’φΨζΒΡ«μ÷§≈ύ―χΜυ÷–ΖΔœ÷ΝΥ“ΜΕ‘Ε‘”≥“λΙΙΧε(phenylahistin), ÷°Κσ Fenical Β»…Η―ΓΖ÷άκ≥ωΨΏ”–“÷÷ΤœΗΑϊ÷ήΤΎΒΡΜν–‘Μ·ΚœΈο Halimide. Halimide Ω…“‘Ής”Ο”ΎΈΔΙήΒΑΑΉ…œΒΡ«οΥ°œ…ΥΊΫαΚœΈΜΒψ, ΙΈΔΙήΫβΨέ. ΧεΆβ ‘―ι±μΟς, ΗΟΜ·ΚœΈοΩ…“÷÷Τ÷ΉΝωœΗΑϊ…ζ≥Λ, Μν–‘ΫœΗΏ. ΫαΙΙ…œ, Halimide(ΆΦ 8) «”… L-±Ϋ±ϊΑ±ΥαΚΆ(Z)-“λΈλΕΰœ©Μ·ΒΡΆ―«βΉιΑ±Υα≤–ΜυΉι≥…ΒΡΜΖΥθΕΰΑ±Υα(DKP)ΒΡ―ή…ζΈο. ‘ΎΕ‘ HalimideΫχ––Μν–‘”κΫαΙΙΙΊœΒΖ÷Έω ±ΖΔœ÷, DKP ΧαΙ©ΝΥ”Ο”Ύ”’ΒΦΩΙΈΔΙήΜν–‘ΒΡ–¬–Ά‘”ΜΖΚΆœύΕ‘«ΉΥ°ΒΡΡΘΑε, ≤Δ«“ L-±Ϋ±ϊΑ±ΥαΓΔΗ’–‘ΚΆΆ®Ιΐ DKP ”κΏδΏρΜΖ÷°ΦδΒΡ«βΦϋ–Έ≥…ΒΡΤΫΟφΦΌ»ΐΖ÷ΫαΙΙ“‘ΦΑΏδΏρΜΖΒΡ 5ΈΜ…œΒΡΌ…ΕΰΦΉΜυΫαΙΙΕ‘ Halimide ≤ζ…ζΫœΗΏΒΡœΗΑϊΕΨ–‘ΨΏ”–÷Ί“ΣΉς”Ο, Ά§ ±ΈΣΚœ≥…HalimideΒΡ―ή…ζΈοΧαΙ©ΝΥΥΦ¬Ζ[76]. ‘Ύ Halimide ΒΡ―ή…ζΈο÷–, plinabulin “―Ϋχ»κΝΌ¥≤ΫΉΕΈ[77].
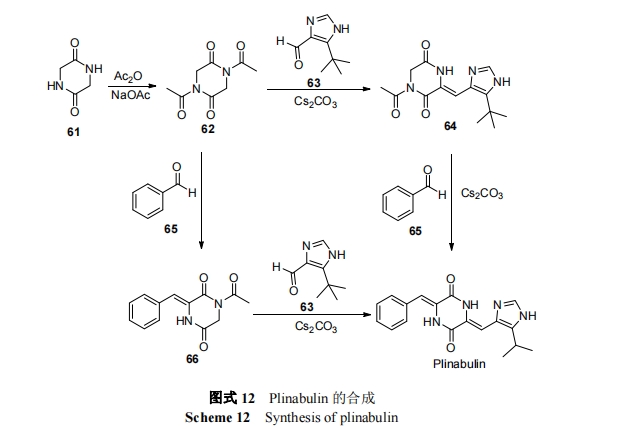
Plinabulin (NPI-2358)”… Beyondspring ΙΪΥΨ―–ΖΔΉςΈΣΕΰœΏΜ·ΝΤ“©ΈοΫαΚœΕύœ©Ήœ…Φ¥Φ÷ΈΝΤΆμΤΎΜρΉΣ“Τ–‘Ζ«–ΓœΗΑϊΖΈΑ©ΜΦ’Ώ. ΡΩ«Α¥Π”ΎΝΌ¥≤IIIΤΎ ‘―ιΫΉΕΈ. ΥϋΒΡΉς”ΟΜζ÷Τ”κHalimideάύΥΤ, ―Γ‘ώ–‘Ής”Ο”ΎΡΎΤΛΈΔΙήΒΑΑΉ«οΥ°œ…ΦνΫαΚœΈΜΒψΗΫΫϋΒΡ ΠΝ-ΚΆ Π¬-ΈΔΙήΒΑΑΉ÷°ΦδΒΡ±ΏΫγ«χ”ρΫαΚœ[78]. ΧεΆβ ‘―ι±μΟς, Plinabulin Ε‘Ης÷÷»Υάύ÷ΉΝωœΗΑϊΨΏ”–Μν–‘, ≤Δ«“Ε‘Ης÷÷Εύ“©ΡΆ“©(MDR)ΤΉΒΡ÷ΉΝωœΗΑϊœΒ“≤ΨΏ”–Μν–‘. ‘ΎΧεΡΎ, Plinabulin Ω…Η…»≈―ΣΙήΡΎΤΛΒΡ’ΐ≥ΘΙΠΡή, ΙΡΎΤΛœΗΑϊΙ«ΦήΈ…¬“, “÷÷Τ÷ΉΝωœΗΑϊ―Σ“ΚΒΡΝςΆ®[79]. Plinabulin ΒΡΚœ≥…Ω…“‘Ά®ΙΐΝΫ÷÷ΆΨΨΕ, ≤ν“λ‘Ύ”ΎΜ·ΚœΈο62”κ5- εΕΓΜυΏδΏρ-4-ΦΉ»©(63)ΚΆ±ΫΦΉ»©(65)Ϋχ––ΥθΚœΖ¥”ΠΒΡΥ≥–ρ(Scheme 12)[80].
“Μœν II ΤΎΝΌ¥≤ ‘―ι, Ε‘±»ΝΥ Plinabulin (N)ΝΣΚœΕύœ©Ήœ…Φ¥Φ(D)÷ΈΝΤΚΆΒΞ”ΟΕύœ©Ήœ…Φ¥Φ(D)÷ΈΝΤΖ«–ΓœΗΑϊΖΈΑ©NSCLC ΜΦ’ΏΒΡ–ßΙϊ. ΫΪ 172 ΟϊΜΦ’ΏΥφΜζΖ÷≥…ΝΫΗω≤ΜΆ§ΦΝΝΩΥ°ΤΫΒΡ¥σΉι, 163 Ηω÷ΈΝΤΉι, ΒΎ“Μ¥σΦΝΝΩΉι”… 30 Ηω–ΓΉιΙΙ≥…, Ηχ“©ΦΝΝΩΈΣ 50DN, 55D; ΒΎΕΰ¥σΦΝΝΩΉι”… 20Ηω–ΓΉιΙΙ≥…, Ηχ“©ΦΝΝΩΈΣ 40DN, 18D. ‘ΎΒΎ“Μ¥σΉι÷–, Ι”Ο DN ΝΣΚœ÷ΈΝΤΒΡΉή…ζ¥φΤΎΈΣ 8.7 Ηω‘¬, ΒΞΕά Ι”Ο D÷ΈΝΤ « 7.5 Ηω‘¬; DN ΉιΒΡΜΚΫβ¬ ΈΣ 14%, D ΉιΈΣ 14.5%; DN ΉιΒΡ“©–ß≥÷–χ ±ΦδΈΣ 12.7 Ηω‘¬, D ΉιΈΣ 1.5 Ηω‘¬; ΒΎ“Μ¥σΉι÷ΈΝΤ–ßΙϊ”≈”ΎΒΎΕΰ¥σΉι. ¥Υ¥Έ ‘―ι÷–≥ωœ÷Ήν≥ΘΦϊΒΡ ADR ΈΣΕώ–ΡΓΔΤΘάΆΓΔΗΙ–ΚΓΔ±ψΟΊΚΆ―α ≥. ‘ΎΝΫΗωΗχ“©¥σΉι÷–, DN Ήι÷––‘ΝΘœΗΑϊΦθ…ΌΖΔ…ζ¬ ΫœΒΆ, ―œ÷ΊADR ΖΔ…ζ¬ ΫœΒΆ. ¥Υ ‘―ι±μΟςΨΏ”–¥σΒΡΖΈ÷ΉΝω(ΘΨ3 cm)ΚΆ÷°«ΑΨ≠Ιΐ±ξΉΦΜ·ΝΤ(ChRx)ΒΡΜΦ’Ώ‘Ύ DN Ήι¥φΜνΦΗ¬ ±»D ΉιΗΏ[81].
6 »μΧεΕ·ΈοΜν–‘κΡ
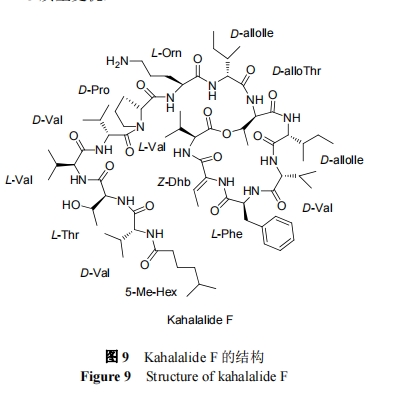
6.1 Kahalalide F
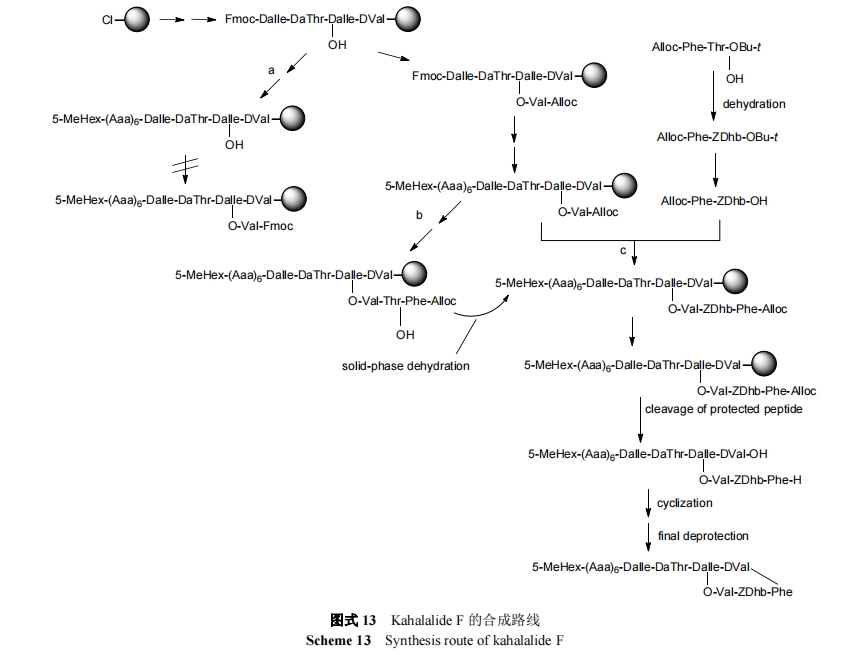
Kahalalide F(ΆΦ9) τ”ΎKahalalideΦ“Ήε, ¥” ≥≤ίΒΡ»μΧεΕ·Έο Elysia rufescens ΚΆ¬Χ‘ε Bryopsis sp ÷–Χα»Γ≥ωά¥. Kahalalide F «Ζ÷Ή”ΝΩΈΣ 1478 ΒΡ °»ΐκΡ, ”…¥σΖ÷Ή”«χ”ρΚΆœΏ–‘«χ”ρΉι≥…, ‘Ύ N-Ρ©ΕΥΚ§”– 13 ΗωΑ±ΜυΥαΚΆ 5-ΦΉΜυΦΚΥα, ΜΙΑϋΚ§ Z-ΕΰΆ―«βΑ±ΜυΕΓΥα(Dhb)[82]. L®°pezMaci®ΛΒ»[82b]±®ΒάΝΥ Kahalalide F ΒΡΙΧœύΚœ≥…ΖΫΖ®(Scheme 13), ΤδΚœ≥…≤Ώ¬‘ « ΙΝ¥‘ΎΙΧœύ…œ―”…λ, ±Μ±ΘΜΛΒΡκΡ‘Ύ ς÷§…œΝ―Ϋβ, ΥφΚσΜΖΜ·, Ήν÷’‘Ύ»ή“Κ÷–»Ξ±ΘΜΛ. ‘Ύ≤Ώ¬‘ a ÷–, Φ”…œ Fmoc-Val-OH ≤ΔΟΜ”–ΒΟΒΫΫœΚΟΒΡ≤ζ¬ , ΆΤ≤β «”…”ΎκΡΝ¥ΒΡ ηΥ°–‘ ΙΤδ”–άϊ”ΎΝ¥ΦδΨέΦ·. “ρ¥Υ, ΫαΚœ±ΘΜΛΒΡVal ”ΠΒ±‘Ύ«ΑΟφΒΡ≤Ϋ÷η(≤Ώ¬‘ b ΚΆ c)÷–. ≤Ώ¬‘ b ΚΆ c ΕΦΩ…“‘ΒΟΒΫάμœκΒΡ≤ζΈο, ΒΪΆ®Ιΐ≤Ώ¬‘ b ΥυΒΟΒΫΒΡ¥÷≤ζΈοΒΡHPLC ÷ ΝΩΗϋ”≈.
‘ΎΧεΆβ, Kahalalide F Ής”ΟΒΡΖΔΜ””κ ErbB3 ΚΆ Akt –≈Κ≈”–ΙΊ[83]. ΧεΆβ Β―ι±μΟς, Kahalalide F Ε‘«ΑΝ–œΌΑ©ΚΆ»ιœΌΑ©œΗΑϊœ‘ Ψ≥ωΫœ«ΩΒΡœΗΑϊΕΨΜν–‘, IC50 ΖΕΈßΈΣ 0.07 µmol•LΘ≠1 (PC3)ΓΪ0.28 µmol•LΘ≠1 (DU145, LNCaP, SKBR-3, BT474, MCF7). ÷Ί“ΣΒΡ «, »Υάύ’ΐ≥ΘœΗΑϊ(MCF10A, HUVEC, HMEC-1, IMR90)Ε‘“©ΈοΒΡΟτΗ––‘ΒΆ 5ΓΪ40 ±Ε(IC50ΘΫ1.6ΓΪ3.1 µmol•LΘ≠1). Kahalalide F ‘ΎœΗΑϊΡΎΖΔΜ”Ής”Ο ±, ΜαΖΔ…ζ“ΜœΒΝ–ΒΡ±δΜ·: œΗΑϊ÷ ÷Ή’ΆΚΆΩ’≈ίΜ·, ΡΎ÷ ΆχΒΡά©’≈ΚΆΡ“≈ί–Έ≥…, œΏΝΘΧεΥπ…ΥΚΆ÷ ΡΛΤΤΝ―. œΗΑϊΚΥΡΎ≤ΜΙφ‘ρ»Ψ…Ϊ÷ ΨέΦ·≥…–ΓΒΡΩιΉ¥, Εχ»Ψ…Ϊ÷ ¥”ΤδΥϊΚΥ”ρœϊ ß, ΒΪΚΥΑϋΡΛ±Μ±ΘΝτ, ΟΜ”–Φλ≤βΒΫDNAΫΒΫβ[84].
ΈΣΤάΦέ Kahalalide F ÷ΈΝΤΆμΤΎΕώ–‘ΚΎΥΊΝωΜΦ’ΏΒΡ–ßΙϊ. ‘―ι―Γ‘ώΝΥ 24 άΐΖϊΚœΧθΦΰΒΡΜΦ’Ώ, 1 ¥Έ/÷ήΉΔ…δΗχ“©. 14 άΐΜΦ’Ώ”–Μ·ΝΤΜρ…ζΈο÷ΈΝΤ Ζ, ΒΪ ΒΧεΝωΒΡΝΤ–ßΤάΦέ±ξΉΦ(RECIST)ΤάΦέΫœ≤ν. ‘Ύ 5 ΟϊΜΦ”–ΤΛΖτΚΎ…ΪΥΊΝωΜ·ΝΤΜΦ’Ώ÷–, Φ≤≤ΓΈ»Ε®ΈΣ 3 Ηω‘¬, ÷–ΈΜΈόΫχ’Ι…ζ¥φΤΎΈΣ 1.7Ηω‘¬(95% CI, 1.2ΓΪ1.9 Ηω‘¬), ÷–ΈΜΉή…ζ¥φΤΎΈΣ 10.8 Ηω‘¬(95% CI, 5.0-…œœόΈ¥¥οΒΫ). ΜΦ’ΏΖΰ“©ΤΎΦδΉν≥ΘΦϊΒΡ…ζΜ·÷Η±ξ±δΜ· «ΉΣΑ±ΟΗ(ALT/AST)ΚΆ ΠΟ-Ι»Α±θΘΉΣ“ΤΟΗ(GGT)ΒΡΖ«άέΜΐ‘ωΦ”. ‘Ύ―–ΨΩΤΎΦδΜΦ’ΏΈ¥≥ωœ÷ΑΉœΗΑϊΦθ…ΌΚΆ―Σ–ΓΑεΦθ…Ό. Kahalalide F «“Μ÷÷Α≤»Ϊ–‘ΝΦΚΟΒΡΜ·ΝΤΈο÷ , ΒΪ”…”ΎΕώ–‘ΚΎΥΊΝωΜΦ’Ώ»±ΖΠΩΆΙέΜΚΫβ, ΗΟ ‘―ι‘ΎΒΎ“ΜΫΉΕΈΚσΆΘ÷Ι[85].
6.2 Elisidepsin
Elisidepsin « Kahalalide F ΒΡΚœ≥…―ή…ζΈο, ”…PharmaMar ―–ΖΔ, ”Ο”Ύ÷ΈΝΤ ΒΧεΝω, ‘χ¥Π”ΎΝΌ¥≤ II ΤΎ, œ÷“―±Μ÷’÷Ι.Κœ≥…Ιΐ≥ΧΈΣ‘Ύ PyAOP ΚΆ DIEA ¥φ‘Ύœ¬, ‘ΎDMF ÷–œΏ–ΆΕύκΡ 67 ”κΜΖΉ¥ΥθκΡ 68 ΒΡ≈ΦΝΣ≤ζ…ζ≤ύΝ¥±Θ±ΘΜΛΒΟΒΫ Elisidepsin. Μρ’Ώ, Ά®Ιΐ‘Ύ CH2Cl2 ÷–ΒΡ DIC, HOBt ΚΆ DIEA ΙΥθκΡ 70 ΜΖΜ·, ΜώΒΟ ή±ΘΜΛΒΡ«ΑΧε 69, »ΜΚσΫχ––Ά―±ΘΜΛΦ¥Ω…ΒΟΒΫ Elisidepsin (Scheme 14)[86]. Elisidepsin ΒΡ–ßΝΠ”κ HER3, ErbB3ΓΔΧ«Μυ…ώΨ≠θΘΑΖΚΆτ«ΜυΜ·÷§ΖΨΥα(”… FA2H ≤ζ…ζ)±μ¥ο”–ΙΊ[87].
ΈΣΤάΦέ Elisidepsin ÷ΈΝΤΆμΤΎΈΗ ≥ΙήΑ©ΒΡ”––ß–‘, Ϋχ––ΝΥ Ib/II ΤΎΝΌ¥≤ ‘―ι. ΫΪ 44 ΟϊΜΦ’ΏΥφΜζΖ÷≈δ÷ΝΝΫΗωΦΝΝΩΉι. Τδ÷– 12 ΟϊΜΦ’Ώ¥Π”Ύ Ib ΤΎΝΌ¥≤ ‘―ι, 32 ΟϊΜΦ’Ώ¥Π”Ύ II ΤΎΝΌ¥≤ ‘―ι. ‘Ύ ‘―ιΤΎΦδ, Ϋœ…ΌΖΔ…ζ ADR, ΒΪ «”…”ΎΝΤ–ß≤ΜΦ―ΟΜ”–Ϋχ––œ¬“Μ≤Ϋ ‘―ι[88].
7 ’ΙΆϊ
ΥφΉ≈ΚΘ―σΩΣΖΔΒΡ≤ΫΖΞΦ”ΩλΦΑΕ‘…ζΈοΜν–‘κΡ»œ ΕΒΡ≤ΜΕœ…ν»κ, ΚΘ―σ…ζΈοΜν–‘κΡ «ΡΩ«Α“©Έο―–ΖΔΒΡ“ΜΗω÷Ί“ΣΖΫœρ. Υδ»ΜΕ‘”Ύά¥Ή‘”ΎΚΘ―σ…ζΈοΒΡΜν–‘κΡ―–ΨΩάζ≥ΧΫœΕΧ, ΒΪ”…”ΎΤδΫαΙΙΒΡΧΊ β–‘Έϋ“ΐΝΥ÷ΎΕύΒΡΩΤ―–ΙΛΉς’Ώ, ΩΣ’ΙΝΥΜ·―ßΚœ≥…ΓΔΙΙ–ßΙΊœΒΓΔ“©άμΕΨάμΓΔΝΌ¥≤ΦΑœύΙΊΒΡ≤ΜΝΦΖ¥”ΠΒ»ΖΫΟφΒΡ―–ΨΩ. ΒΪ”…”ΎΚΘ―σ…ζΈοΜν–‘κΡΫαΙΙΗ¥‘”ΓΔΜν–‘≥…Ζ÷Κ§ΝΩΒΆΦΑΖβ±’ΒΡ N Ρ©ΕΥΒ»ΧΊ βΫαΙΙ, ΙΒΟΗχΩΣΖΔ―–÷Τ‘ωΧμΝΥ÷νΕύάßΡ―. ¥”ΡΩ«Α…œ –“©ΈοΚΆ¥Π”ΎΝΌ¥≤ ‘―ιΫΉΕΈΒΡΜν–‘Μ·ΚœΈοά¥Ω¥, Ε‘”ΎΚΘ―σ…ζΈοΜν–‘κΡΒΡΩΣΖΔ, ÷ς“Σ‘ΎΩΙ÷ΉΝω“©Έο―–ΖΔΝλ”ρ, Μ·Έοά¥‘¥Ψ÷œό”ΎΚΘΆΟΓΔ”σ¬ίΓΔΚΘΟύΓΔΚΘ« ΓΔ»μΧεΕ·ΈοΒ»…Ό ΐΦΗ÷÷…ζΈοΧε÷–, Ψχ¥σΕύ ΐΚΘ―σ…ζΈο…–Έ¥ΒΟΒΫΩΣΖΔ.
ΈΣΫΪΗϋΕύΒΡΚΘ―σ…ζΈοΜν–‘κΡάύΜ·ΚœΈοΩΣΖΔΈΣ“©Έο, Έ“Ο«”Π¥”»ΐΗωΖΫœρΫχ––: (1)“‘Χλ»ΜκΡΈΣœ»ΒΦΜ·ΚœΈο, Ε‘ΤδΫχ––ΙΠΡήΜ·–ό Έ, ―–ΨΩΤδΙΙ–ßΙΊœΒ, “‘…Η―Γ”–ΗϋΚΟ“©–ßΒΡΜν–‘κΡάύΥΤΈο; (2)ΚΘ―σ“©ΈοΒΡ―–ΨΩ–η“ΣΧΫ≤βΓΔœ÷¥ζ…ζΈοΓΔΖ÷άκ¥ΩΜ·ΚΆ≤ζΤΖ÷Τ±ΗΒ»œύΙΊΦΦ θ, Ι –ηΦ”¥σΝΠΕ»Ϋ®…ηΜν–‘κΡΒΡΖ÷άκΦχΕ®–η“ΣΒΡΗΏ–ßΝιΟτΒΡΦΦ θΤΫΧ®. (3)ΚΘ―σ «“ΜΗωΨό¥σΒΡ±ΠΩβ, ΜΙ¥φ‘ΎΉ≈–μΕύΈ¥ΩΣΖΔάϊ”ΟΒΡ…ζΈοΜν–‘κΡάύΜ·ΚœΈο, ”ΠΫχ––ΗϋΕύΒΡΙψΖΚ…ν»κ―–ΨΩ, “‘―Α’“ΗϋΕύΚΆΗϋ”–Φέ÷ΒΒΡ–¬–ΆΚΘ―σ“©Έο.
Οβ‘π…υΟςΘΚ±ΨΈΡΈΣ––“ΒΫΜΝς―ßœΑΘ§Αφ»®Ιι‘≠Ής’ΏΦΑ‘≠‘”÷ΨΥυ”–Θ§»γ”–«÷»®Θ§Ω…ΝΣœΒ…Ψ≥ΐΓΘΈΡ’¬±ξΉΔ”–Ής’ΏΦΑΈΡ’¬≥ω¥ΠΘ§»γ–η‘ΡΕΝ‘≠ΈΡΦΑ≤ΈΩΦΈΡœΉΘ§Ω…‘ΡΕΝ‘≠‘”÷ΨΓΘ
