ժҪ���������µ����������κͷ�������������ҩ��Ŀ����ڹ�ȥʮ����ȡ���˾��չ������ҩ������������β����˻�ѧ�����﷽��������������ӱ����ƺ͵��Ͳ��ԣ��������ڿ˷�����ҩ��Ĺ���ȱ�ݣ����ƶ�������ij�����չ�������Ѿ���ò��о��˶�����Ȼ����������ҩ������˶���������������ܽ�������ҩ��֡����������η����Ŭ���ͳɾͣ��Լ�����Ŀǰ��Ӧ�á����ǻ�����������������ҩ��δ����չ�ļ�ֵ����ս��
������������һϵ����������ɵ�һ�����ҩ�������ͨ��Ϊ500-5000Da���������ĵ��о�ʼ�ڶ��ȵ��ء��߲��ء���ѹ�غʹ����ټ����ͷż���(GnRH)����Ȼ���弤�ؼ������������ض��������ԵĻ����о�[2]����1921��ϳɵ�һ�����������ȵ����������Ѿ�ȡ����������Ŀ�ijɾͣ�ȫ������80��������ҩ���������ˣ�����ҩ��Ŀ����ѳ�Ϊҩ���о��������Ż���֮һ��
20�����ϰ�Ҷ�����Ƿ����˼��ֿ���������������������ģ������ȵ��غʹ�������Ƥ�ʼ��أ���Щ��������Ǵ���Ȼ��Դ���о��ͷ�������ġ��ȵ�����һ�ֺ���51����������ģ����ķ��ֺͿ�������Ϊ��ҩ���������ش��ѧ�ɾ�֮һ������1921����FrederickBanting�״η������������Frederick��CharlesBest��һ������[3,4]�����״η�����һ�����������������ߡ�1923�꣬�ȵ��س�Ϊ��һ����ҵ������ҩ��������츣��ǧ�������ߡ�Ȼ����20���������ȵ��صIJ����������г�����ţ�ȵ��غ����ȵ��صȶ���Դ���ȵ������ȵ����г���ռ��������λ��90�ֱ꣬���������ȵ���ȡ����
20����50�����90����������������DZ�����ļ��ؼ���������̱������ͱ���7��ͬʱ�������ʴ����ϳɡ��ṹ�����Ͳ�����ȡ��ʵ���Խ�չ������������ҩ����з���ȫ�����н�40������ҩ�������ֵ��ע����ǣ�������Ȼ���⣬�ϳɴ߲��ء��ϳɼ�ѹ�ء��������ȵ��صȺϳ���Ҳ��ʼ��������
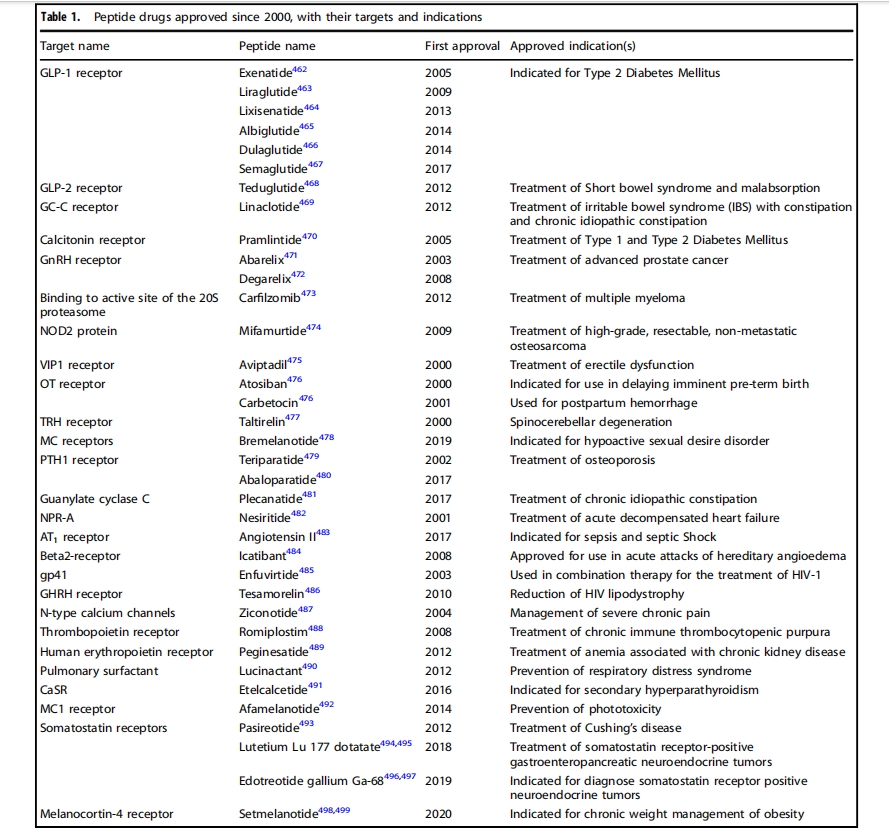
����21���ͣ�����ҩ���з������¼�Ԫ���ṹ����ѧ�����������Ƽ������ͺϳɷ��������Ľ����������˶���ҩ���з����̣��ѽ����˰�������ҩ��֡�ҩ����ơ����ĺϳɡ��ṹ���Ρ��������۵Ȼ��ڵĶ���ҩ���з���ϵ����2000��������ȫ����33�ַ��ȵ��ض���ҩ���1�������⣬��Щ����ҩ���Ѳ����Ǽļ���ģ���������Ȼ��������ɡ����磬����Τ����һ��36��������ķ����ģ�ģ����������ȱ�ݲ�����HIV�����ף�������������HIV-1 ;�뿼ŵ����һ�ִ�����Conusmagus����ȡ�������ģ���2004�������������������������ʹ�����³����һ���ȸ�Ѫ��������2(GLP-2)������������ƶ̳��ۺ�������������DNA��������Ĵ˾��������죻����³���������ȸ�Ѫ��������1(GLP-1)�Ļ�ѧ�ϳ������ͨ����C-16֬���ᣨ����ᣩ��������л��������е�λ��26���ϵĹȰ����������Ӷ��ɣ�����ΪGLP-1���弤����������2������(T2DM)��������Щ����ҩ�ﶼ�����ڹ㷺������������������ơ������ơ���ʹ�ơ������ơ���л�ơ���Ѫ�ܿƺͿ����ơ�����Ϊֹ������170��������ҩ�ﴦ�ڻ�Ծ���ٴ������Σ���2�������и�������ҩ�ﴦ���ٴ�ǰ�о��Ρ�
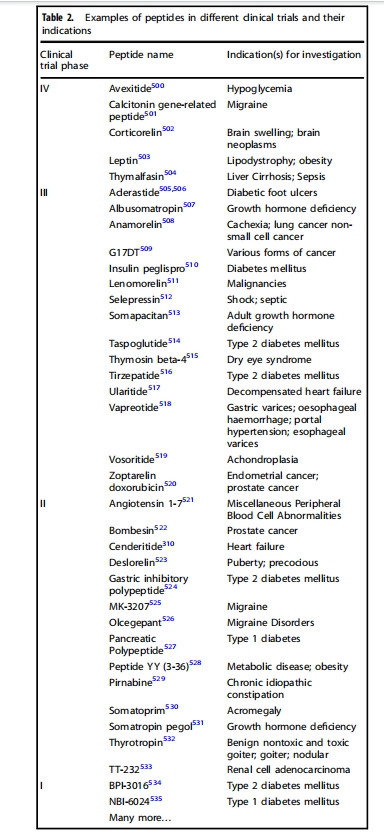
����ҩ����ҽҩ�г���ռ���൱��ı��أ�2019��ȫ�����۶��700����Ԫ����2013�����������ࡣ��Njardarson��ͳ�ƣ�2019�����۶�����ǰ200��ҩ���У���10�ַ��ȵ��ض���ҩ�����˼���ǣ�����ҩ�����۶�ǰ���ľ�Ϊ����2������GLP-1���������������19λ��Trulicity���������ģ������۶�43.9����Ԫ��������32λ��Victoza������³�ģ������۶�32.9����Ԫ��������83λ��Rybelsus������³�ģ������۶�16.8����Ԫ��ͼ1����
���Ļع�������ҩ�����ʷ��չ������ҩ��ֵ����½�չ�������ص��ע�������ĵ�ҩ�����ԣ����ص���ܸĽ�����ҩ����ơ��ϳɡ����κ��������¼�������Ϊ����ҩ���Ӧ���ṩ�µ��ӽǡ����ǻ�������߲�������ļ�ƪ�����Թ���һ���Ķ�.
�������ģ��ŵ��ȱ��
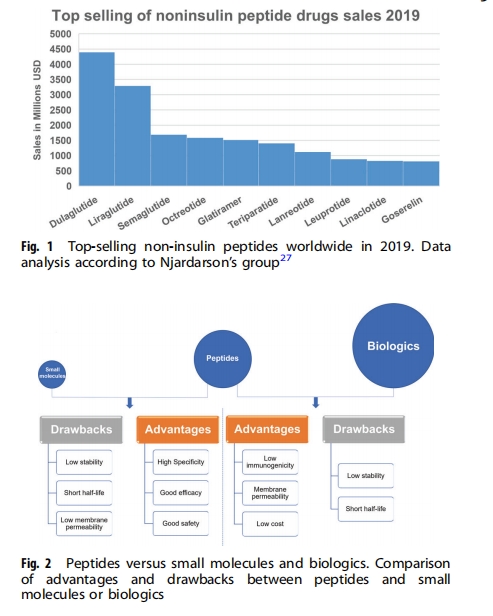
��������ͨ���������ء��������ӡ����ʡ�����ͨ�������Ⱦ����������ϸ�����������ϲ��Ը�����������������ϸ����ЧӦ�����÷�ʽ�������Ƽ������������Ե����ʺͿ��壩���ơ�Ȼ�����������Ƽ���ȣ��������ĵ�����ԭ�Խϵͣ������ɱ��ϵ�.
С����ҩ������ƾõ�������ʷ�����������ɱ������ۼ۸�͡��ڷ���Ĥ���Ժõȹ������ơ����ĺ������Ƽ��������ʻ��壩��ȣ���Ȼ��ȡ�ͻ�ѧ�ϳɵ�С���Ӿ����о����Եļ۸����ơ��ڷ�С���Ӿ��и��õİ�ȫ�Ժ�����������Եĺô���ͬʱ���ǵ�С�ߴ�Ҳʹ�����ܹ���ϸ��Ĥ����ϸ���ڵķ��ӡ�Ȼ�������ǵ�С�ߴ�Ҳ��ζ�����Ǻ�����Ч�����ƴ��������ã����絰����-����������ã�PPI����PPI ͨ��ռ�� 1500�C3000 A2�ĽӴ��������С������������ӳߴ����ޣ�ֻ�ܸ��� 300�C1000 A2�ĵ����ʱ��档���֮�£�����ҩ����ص�������ѧ���ʣ����������ijߴ�����ĹǼܣ�ʹ�����ܹ���Ϊ PPI ��ǿЧ���Ƽ�,С����ҩ����ٴ�Ӧ��Ҳ�������Ե��ڶ�����ҩ����ܵ����ƣ���������������������ᶼ���Ұ��ἤø���Ƽ���������Ѫ����Ƥ�������ӣ�VEGF��������Ұ��ἤø�ṹ����ԣ�������Ѫ���������ã��������ư�֢���ߣ�Ȼ��������Ҳ����������ø���壬��˿����/�հ��ἤø���壬����ϸ������.
��Ϊ������Ȼ����������Ƽ����������ľ�����������ȱ�㣨ͼ2�� ��Ĥ��ͨ�Ժ������ȶ��Բ������ҩ�↑������Ҫ����ʯ.
1�����ĵ�Ĥͨ�Խ���������ҩ���Ĥͨ��ȡ���ڶ������أ��������ij��ȡ���������ɵȡ�����һ��������ϸ��Ĥ����ϸ���ڰе㣬�������������ҩ�↑���е�Ӧ�á�2018��Lau�ȱ������ٴ�������90%���ϵĶ��İ���ϸ����е㣬����G����ż�����壨GPCRs���������ټ����ͷż��أ�GnRH�����塢�ȸ�Ѫ��������1��GLP-1�����塣
2���������ڵ��ȶ��Խϲ��Ȼ����ͨ�����������ӵİ���������ɣ���ȱ�������������ṹ��������ȶ��ԡ���û���κα���������£�����¶�ڻ�����ʱ�������������ױ�����øˮ����ƻ�����Щ���еĻ�ѧ����ʹ���ڻ�ѧ�������ϲ��ȶ�����˥�ڶ̣����������졣
���ĵ���Щ�������������Ƽ�Ϊ����ҩ�↑����������ս��ҲΪ����ҩ�����ƺ��Ż������˻����ͷ���
�������ĵķ�չ·�������֡��������Ż�
����ҩ���з�
�����ڵ���Ȼ��/����
����ҩ���з�����ʷʼ�����þ��г���о����������ܵ���Ȼ���غ���������������ȱ������ļ��������� 1 ������ 2 ��������ȱ������Ѫ��ˮƽ������ȵ��ء��������Ʒ�����ע���ȵ��أ���̼��ȵ��ط�����ذе㣨�� GLP-1 ���壩�����ȵ��ء�Ѱ����Ȼ����ͼ��ػ��ö���ͬԴ����ȵ��ء�GLP-1���������ء�GnRH��8-Arg-��ѹ�غʹ߲��أ�������ǣ���������ҩ���з���������ԣ�ͼ3��)��Ȼ������Щ��Ȼ�������ڵ�ȱ�����������Ƕ��Ż�����Ȼ���е���Ȥ���Ӷ�������һϵ����Ȼ����ģ����ҩ�
����ģ����
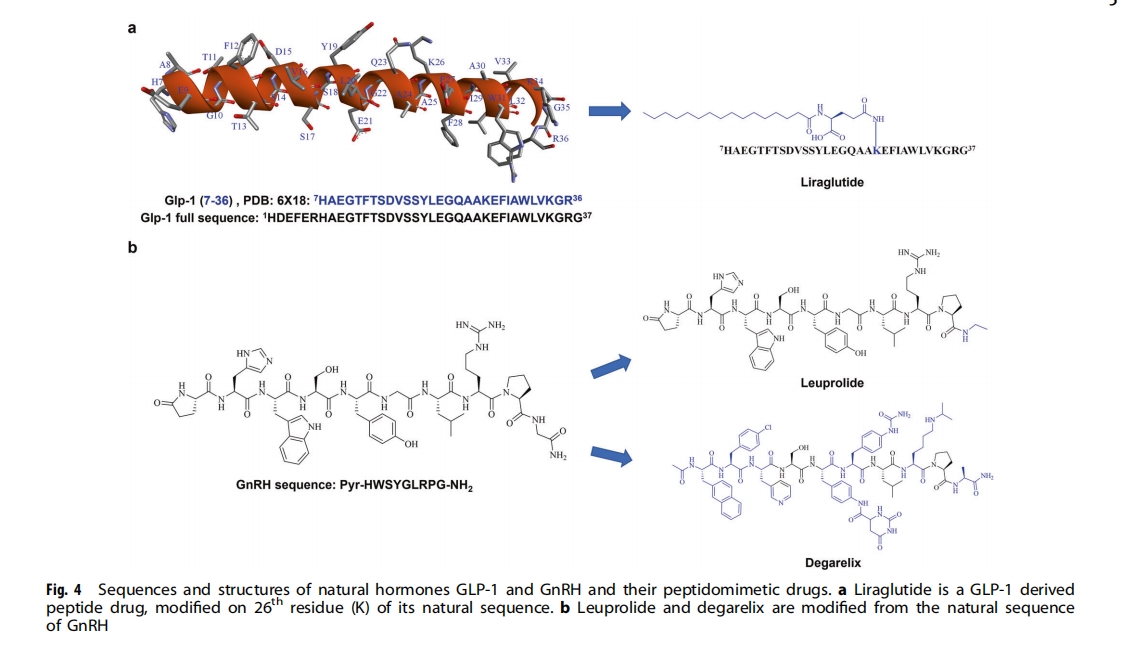
GLP-1�����Ķ���ҩ�ͼ4a����GLP-1��һ��37��������Ķ��ģ��ɵ����ȵ��صIJ����ͷ��ڣ������ڵİ�˥�ں̡ܶ����Ǹ����˴�����Ŭ�����������У�����ǿ���ּ��ص��ȶ��ԣ�ͬʱ������Ч����ҩ�����ã��Ӷ���������������Ŀ�2��������ҩ�Trulicity���������ģ� ��Victoza������³�ģ���Ozempic������³�ģ���
�����ټ����ͷż��� (GnRH) ��������ҩ�ͼ4b����GnRH ��һ�ֺ��� 10 ����������ģ����������е� GnRH ��Ԫ�������� GnRH ��Ȼ���е����ѵ��¼�����ҩ��Ŀ����������������ֺ͵ؼ���ˡ���������ͨ������ GnRH ��������� GnRH ��ͬ��������ԣ������� GnRH ���弤�������������Ƽ��ط�Ӧ��ǰ���ٰ����ӹ���Ĥ��λ֢���ӹ������������졣��Ȼ�ؼ���˵������Ǵ� GnRH �Ż������ģ�����ͨ�������Ե��� GnRH �����ϳ䵱 GnRH������������������ǰ���ٰ���
������������������ҩ��Ҳ��Դ����Ȼ����1�����������ģ�һ����������ģ������ҩ��������Ʋ����������ص������ʹ�������ȥ����ѹ�أ�һ�� 8-Arg-��ѹ��ģ������ҩ������������֢��ҹ��֢�����������أ�һ���������Ʊվ��Ĵ߲���ͬԴ��Ͱ������࣬һ��������������Ĵ߲���������
����Ȼ�����м�������
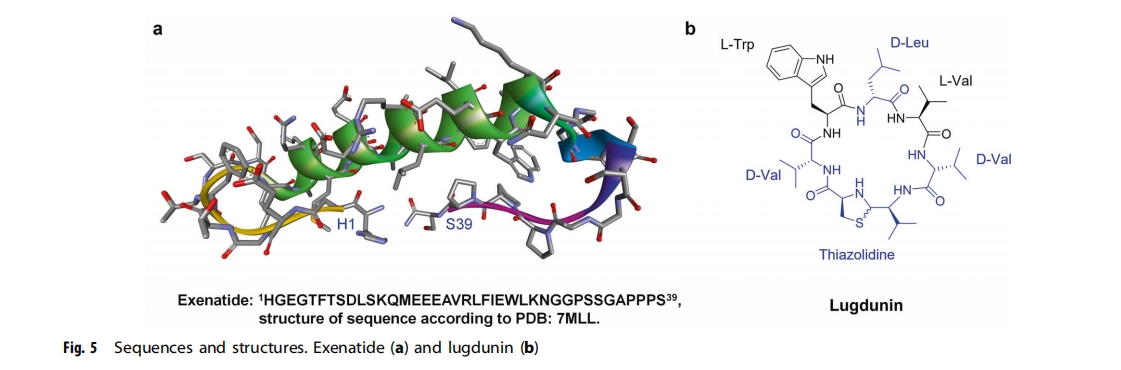
����ϸ���������ֲ��Ͷ����������������Ķ������������ԣ������߶���������Ϊ��Ѫ����Ƥ��������(VEGF)�����VEGF-F��svVEGF������ͨ���Ƕ�����ḻ�Ļ��ģ��л�������80������ͨ����������ͨ��������Ĥ��������յ�ϸ�����ԡ������ߺ�Ы�ӵĶ�Һ���ѱ�������������Ӧ�á����⣬��Gila���ﶾҺ���Ż��İ������ģ�ͼ5a����һ��GLP-1�����������뿼ŵ����һ���������ݵĶ�Һ�ģ��ѱ�������������������ʹ
�Ǻ������ģ�NRP���Ǵ���Ȼ�����м�������һ���ġ������к��еķDZ��л���ζ��NRP����ͨ����ͳ�ĺ���������ϳ�;�������ģ������ɷǺ������ĺϳ�øͨ������ʼ���ӳ�����ֹģ����ɵ�;�������ġ��������ϳɵ�����ȣ�NRP��ˮ��ø�ĵֿ�����ǿ�������ڵ��ȶ���Ҳ���ߡ��о�����NRP��Ҫ��Դ��ϸ����������������ù�ء����߾��ء�·��ù�ء���ͼ5b)�����п������Ե�������Ͷ����Լ����п��������Ե�a-���ަ���������A�ͷ��߾���73��74 �����⣬�����ǻ�״�ģ�����ͨ����ֲ���м�����һ���ض�NRP 75��76��77 �������������B�Ͱ�Ī����˹��78��79����Щ��ҩ����ֳ���ǿ��Ѫ���ȶ��ԣ�ʹ���ܹ��ڷ���ҩ��Ȼ����NRP�ĺϳɺ�Ч��ϵ�о���NRP�����ս�Ժ��������˷ܵ��о�����֮һ��
���ݵ�����-����������ú��������
��������ѧ�ͽṹ����ѧ�ķ�չ���·����������������ϸ�����̺����﹦�ܵĵ�����-�����������(PPI) ������Ϊֹ�����о��˳���14,000��PPI����ռ��������PPI��1%���ҡ�PPI���������༲���е�������Ҫϸ��ͨ·�������DZ�ڵ�ҩ��е������ΪPPI���Ƽ�������С���ӡ��������Ⱦ����������ƣ���˻�����֪PPI����ṹ��չ��һ���µĶ���ҩ��ּ��������ĵĺ�����ƣ�����Ϊ��һ�ֺ���ǰ;�ķ������Ͷ���ҩ���ѡ��IJ��ԡ�
�ĵĺ�������漰����Ŀ��PPI��������ṹ�ļ��������������Ϣѧ������PPI��Ͻ����������Ϣѧ�ͼ����������ʶ����������õ����ʱ���ı��谱���ᡣ��Щ���谱���ṱ����PPI����Ҫ����˹�ܣ�ͨ����Ϊ���ȵ㡱 ���ȵ�����ǵ����ʵ�����Ƭ�Σ�Ҳ�����ǵ����ʲ�ͬ�����ṹ�ϵķ�ɢ�л���PPI�ĵ��ڼ�����ƻ�����Щ�ȵ㣬Ҫôֱ��ʹ������Ƭ�Σ�Ҫôʹ�ý���ɢ�л�����������Ϊ��ʼ���еIJ��ԡ�Ȼ����Ϊ���������Ժ�������ѧ���ʣ���Ҫ��һ�������ĵĿ����ͽṹ�Ż��������Ļ������������Ρ����磬ͨ���о��ṹ-���Թ�ϵȷ�������IJл�������DZ���л���������Լ������н��л�ѧ�������ȶ��Ķ����ṹ������ת�ǡ����������к����칹������ǿ������Բ�����������ѧ���ʡ�
ͨ���ɾ���չʾ���������ѡҩ��
�ɾ���չʾ��һ�ָ�Ч���ɿ��ļ���������ʶ������б�����壬�� Smith �� 1985 ���״α������ɾ���չʾ�������鼼�����ɾ��������ưб����塣�ɾ�����ֻ�������е����ʰ�������ģ������� NRP�����ָ�ͨ������������ʶ��ҩ������������������ġ��ɾ���չʾ�ѹ㷺���ڷ����µ������塣Lerner ���˱���ͨ���ɾ���չʾ������ GLP-1 ������Ĥ���������ǿЧ����������������ʡ��ĺͶ�Һ����Ҫ��Ϊ�����������⣬����ת���������� (TGF)-��1 ���Ƥ������������ (EGFR) ���ģ��Լ��ƻ�����άϸ���������� (FGF)-1-FGFR1 ����õ���������ͨ���ɾ���չʾ���ֵ���ҩ����������ӡ��ɾ���չʾ���������·�չ������Ѱ�Ҹ���Ч��ɸѡ�������Լ��������е�����ѡ������ͨ�������ɾ�����ѡ���ڡ�Heinis �������á��ɾ����ϡ����μ����Ӵ�ͳ�ɾ���չʾ�л�û�ѧ�����ģ����˫���ѻ��ġ���һ�ֲ����漰������ӱ��չʾ���������磬Schumacher ���˿����˾����ɾ���չʾ��̽�� D �����ģ��� Szostak ���˽��� mRNA չʾ�Է��ֺ�ѡ����з���Ȼ������Ĵ��ġ�Suga �������ú�����չʾ���������� D ������ͷ���Ȼ��������ȵ��ģ�����������Դ���.��Щ��չʹ�������ܹ�����������չʾ�Ŀ⣬�Է����µĺ�ѡ�ġ�
�������ĵĺϳ�������
���־�������DZ����������ҩ�↑���ĵ�һ����֮��ͨ����ѧ������ϳ��IJ��������������Ը�����ҩ�����ԡ����������ܽ������������������õ��Ļ���������
�ĵĻ�ѧ�ϳ�
�ĵĻ�ѧ�ϳɼ����Ѿ��ܷ���ر���1963��Merrifield�����Ĺ����ĺϳɣ�SPPS������.SPPS�����ڷ����ۺͺϳɲ��Ϸ���õ��������Ľ��������ִ����������з�����������Ҫ�����á���ͨ����������ż�����ѱ��������һ���ķ�Ӧ�������ٽ����ĵĺϳɣ����һ���������Զ����ĺϳ��ǵķ����������鼼����ȣ�SPPS��õĴ��ĸ��ӵ����������������ﻯ�����ø��DNA��RNAƬ�Ρ�����صĵ����ʺͶ��ġ����⣬����SPPS��Ʒ�е����ʺ�����ʶ����Ϊ������Ҫ���Ժϳɹ����еIJ���ȫ��Ӧ��Ӧ��ʹ�ú����Ĵ�����Լ�
SPPS��������������Ȼ�ż��������ۺ�����֬�ϣ����������ӱ��������ͷų�����һ��ѭ����ͼ6����������֬����4-����������(HMBA)��֬��Wang��֬��2-����������(CTC)��֬��Merrifield��֬�����ڽ������������Ȼ������ĵ�C�ˡ��ִ��Ĺ�ҵͨ������֬�벻ͬ�Ľ�ͷż���������˸��ֹ�������֬���Ӷ��ܹ��ڹ����кϳɳ��ĺͽ����Ļ������ںϳɹ����У�������İ����Ͳ���ͨ���ܵ���ͬ��ѧ���ŵı�������ᵼ���ľۼ������ʹ��ĵĴ��ȡ�Ŀǰ�ѿ�����������Ҫ��SPPS���ԣ�Fmoc-SPPS��Boc-SPPS���ֱ�����ȥ����Ҫ�İ��������ţ����̼����ʻ���Fmoc�����嶡���ʻ���Boc ����
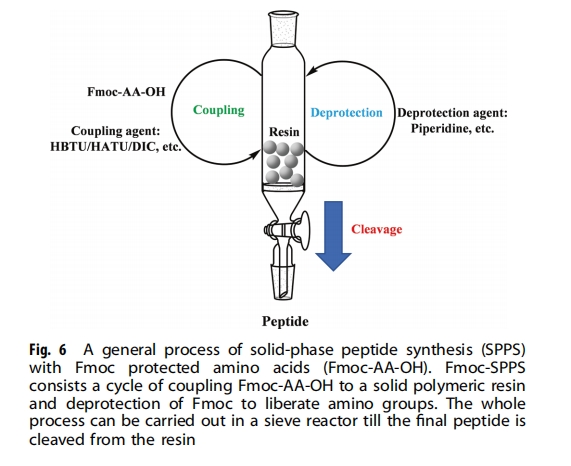
Boc-SPPSʹ������������Һȥ������������ʹ���������Һ�ѽ����յ��ģ�����Щ���̰����Ŵ̼�����ζ�Ͷ��ԡ�Fmoc�����ڸ��º͵�������ȥ�������ͨ����ѡFmoc-SPPS���ԡ�Ȼ����Boc-SPPS���ڳ��ĺϳ������ƣ���Ϊ���������ѱ���������Ч�ƻ��ĺϳɹ����еľۼ���Fmoc-SPPS�о�Ŀǰ��Ҫ�����ڽ��������Ҫ���⣬�������ĺϳɹ����еľۼ���ijЩ���е��춬���ǰ����γɡ��Ѳ��ö��ַ���������Ӧ�õ�ȡ������֬���������������ٷ�Ӧʱ�䡢ʹ�û���ܼ���Ϊ��Ӧ��Һ���Լ�ʹ�üٸ�������������H���Ա�������SPPS�����еľۼ���Fmoc-SPPS�������춬���ǰ����γ����������˴��ĵĴ��ȡ������춬���ǰ��γɵĽ��������ʹ�������ٷ�Ӧʱ�䣬��ʹ��N-��-���Asp�CGly���� �������ѱ�������������1-�ǻ���������HOBt����OxymaPure��
Fmoc-SPPS�ϳ�����50���л�������Գ��棬���ϳ��ģ�>50�������ᣩ�Ļ�ѧ�ϳ���Ȼ������ս�ԣ��������ڴ��ģ�����С�ʵ���ҹ�ģ���ĺϳ����������ִ��Զ��ĺϳ��ǣ���CEMLiberty PRIME��CSBio II���Զ����С���Щ�����Զ��ĺϳ��ǿ����ṩ���192����ͬ���е�˳��Ͷ�ƽ���ĺϳɣ�ʹ�ú���������������̷�Ӧʱ�䣬��ʱʹ�����������ȷ���ѱ������̡�����ϳ�������ʵ���ҹ�ģ���ĺϳɼ�Ϊ���ã����Կ�������������ģ��Թ���һ���Ľṹ�����о���Ȼ��������ȱ�������豸�Ͳ����ȵĹ��ȣ�������������ڴ��ģ�������е�Ӧ�����ޣ�����ܵ��¸�����IJ�������ˣ���������������淶(GMP)�������º͵ķ�Ӧ����������ȵؼ��ٸ���Ӧ��������ʣ���˴��ģ�������ģ�>50�������ᣩ��Ȼ������ս�ԡ�
��ѧ�ĺϳɼ����ķ�չ��������SPPS�����ķ�չ�����������������ĵĿ�����һЩ������ҩ���߲��غ��������ģ����û�ѧ�ϳ�����������ҩ��ɷ֡��ĵĻ�ѧ�ϳɻ������������ǧ�������Ρ�
�ĺ���ģ����Ļ�ѧ����
������Ϊһ�����������ҩ�������������仯ѧ�ṹ������أ����ĺϳɺ���Ҫ����ҩ�ﻯѧ�����������Σ�ģ�⡢�ȶ�������Ķ����ṹ���������������ԣ�ʵ�ֶ���ҩ���ѡ���ԡ��ȶ��Ժ��ܽ��ԡ�
�������ȵ��ĺ�ѡҩ��֮ǰ������ȷ��������������ѧ���Ե���С�������С���������ɨ�裬��Ϊ������ɨ�裬ͨ�����ڽ�ÿ���л��滻Ϊ�������Բ���һϵ���ȵ����������ȷ����Щ�ؼ��л������ȵ��ĵ�������ԣ����Խ��ͱ������滻�IJл�����Ҫ�������Բ��������ͱ������滻�IJл��Ƕ���ġ�Ȼ����ȵ��ĵĿ��滻�л��� C �˺� N �˽��н�һ�����Σ��Բ������յ���ҩ�
�ĹǼ�����
�Ǽ����ε���Ҫԭ��֮һ������ĵĵ���ˮ���ȶ��ԡ����еĵ���ˮ��λ�����ͨ���ȶ����о��ʹ�л��ⶨ��ʶ�𡣹Ǽ����ΰ����� D-������ȡ�� L-�����ᡢ����������ᡢ�Լ����� ��-������140������ , ����Щ����Ȼ���������������У��ر����ڵ���ˮ��λ�㣬���ӳ�����ҩ��Ѫ����˥�ڵ���Ч���ԡ�һ���ɹ���������������ѹ�أ�����Դ�ڼ�ѹ�أ��������Ƶİб�ѡ���ԣ���Ѫ����˥�ڸ�����
�ĵIJ�������
�ĵIJ������������ĺϳɹ����������������滻��Ȼ�����ᣬ��������������Ͱб�ѡ���ԡ���Ȼ������������ı��壬��߾����ᡢ�����Ұ���� ��-�������ᣬͨ�������۵ģ����ҿ��Է�����������ĺϳɹ����ж��IJ������л�ѧ���Ρ����� GLP-1 ������ҩ�������³�ĺ�����³�ģ��������εIJ�����
ͨ�������Ͳ�������ģ����ȶ������ṹ
���е�����������������»����ͷ�������ˮ����ò������γ��ȶ��Ķ����ṹ������ˣ���Ҫ��������N �˻� C �˻�������ж�������Σ���ģ����Ȼ����Ľṹ�� PPI �е��ȵ㣬���ȶ������ṹ���Բ�����ǰ;������ҩ���ѡ�
�Ļ�����������һ�ֳ����������μ��������������ֲ��ԣ�����ͷ��β�������Բ����Ͳ����Բ���������ͼ7�����Ļ����������ӵ���ˮ���ȶ��Ժ�ϸ��ͨ�ԣ�������ģ����ȶ��ĵĶ����ṹ����������������������������ӣ��Ͳ����γɻ���ת�ǽṹ��������ͨ��Ԥ����֯����������ôٽ���Щ�����ṹ���γɡ��Ļ���Ҳ�������ȶ����������ṹ���� ��-������ ��-�۵���
��ģ���-�������ȶ���������������ĵ����ʶ����ṹ����֮һ��Լռ���е����ʽṹ��30%-40%����-�����ɷ���������γɣ�ռ�����ṹ��90%��ģ�����е� �� �������Լ��� PPI �ĵ��ڼ�������ͨ�����������������ù��ۼ�ȡ���������Ϊ�������HBS�����ȶ� �� �������� �� �����ṹ�У�λ�� i��i + 4 �� i +7 �İ��������λ��ͬһ�࣬ͨ�� i �� i + 4 �� i �� i +7 ��������������Ч�ӽ�����ԭ�Ӳ��������������ṹ���γ���������Ǹ����˾�Ŭ�����о���ͬ�Ľ�������������������Ľ�����ͼ7)��ͨ���Ȱ�����춬����������������γ��������ţ�ͨ���ð��װ����ͬ�Ͱ��װ����滻�л��γɶ�������Լ�˫�������ӡ�����������������һ���µĽ��������������ȶ���-�����ṹ��ʹ�÷���Ȼ�簱�����滻i��i + 4��i��i +7λ�õIJл��������˽����γ����ӡ�HBS���β������ù��ۼ�ȡ����-�����ĵ�һ���������Ԥ����֯�����ṹ��Cabezas��Satterthwait����ʹ���¼�����HBS����ģ���-������Arora�о�С��Ҳ��HBS�Ľ����˹㷺���о���ʹ��ϩ�����������ȶ���-���������������ʼʹ��HBS�������ȶ���-���У��Լ���Щ�����ĵ�������ԣ�������ǰ�ڹ�����Ҳ������HBS�����β��ԣ��ص������һ��������SPPS;�����Լ�HBS�ڦ�����ģ����ȶ��е�Ӧ�á�
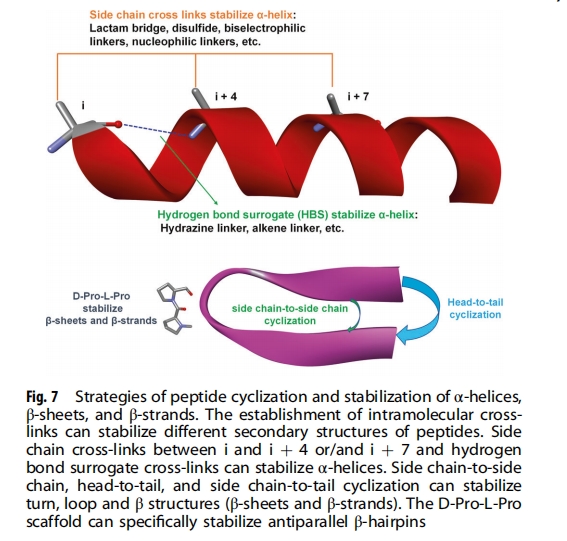
�� ���� �� �۵�����ģ�⡣�� �۵��� �� ��������һ�����ת��ģ��ĵ����ʶ����ṹ�����������ȶ� �� �۵�ͨ����ͨ������ D-�����ᣨ�� D-Pro �����������γ�ת�ǽṹ��ʵ�ֵġ�D-Pro-L-Pro ģ����������֪��֧�ܣ������ȶ����ֳɹ��� PPI ���Ƽ��еķ���ƽ�� �� ���� ��������������צ� �۵�ģ����Ҳ�����ڴ��� �� �۵��� �� ���ṹ��
��ѧ�������Ʊ�����ṹ�Ķ������������Ч���������ȶ��Ժͻ��Ե������ʹ���ֶ���ҩ������ٴ��������ּ�ѹ�ء�����³�ġ�����³�ĵȡ�Ȼ����Щ���β�����ͬʱ��ߵ���ˮ���ȶ��Ժͻ��ԣ��������D-������ͨ���������ӳ����ĵ�Ѫ����˥�ڣ���D-���������εĶ��ĺ��ٱ��ֳ���Ч��������ԡ�
���鼼��������
��ѧ�ϳ��ǹ�ҵ�Ʊ��ĵ���ѡ��������Ϊ����������������ʰ�����֮��Ķ�ܺϳɽṹ��Ԫ���������Ȼ���������������������̽�룬�Ӷ�������һ�����λ��ϡ����⣬��ѧ�ϳɹ��̿�����ȫ�Զ������������������ģ����Ϊ�������ĺ������ṩ��һ�ַ�����Ч�ķ����������ĵĻ�ѧ�ϳ���Ȼ������ս�ԣ������Ҫ������ԡ�
���˻�ѧ�ϳ��⣬�������Ļ�����ͨ���������﷽���Ʊ�������ͨ����ȡ����Ȼ��Դ������������ġ�ø�ϳɡ����͡����� DNA �����Ͱ�ϳɡ���Щ�������Ե���Ӧ�û����Ӧ�ã�����ȡ�����Ʊ��ĵĸ����Ժ��Ѷȡ�
����Ȼ��Դ��������ҩ���ʵ�������ݵ�20����20�������ʱ�ȵ����״δ����������з���������������������������ʮ���˵��������ȵ��صĿ����Գɹ����¹��ڶ����Ʒ�������������ǣ�����������ֶ���Դ����ҩ��ɹ������ٴ�ʹ�ã������������Ƥ�ʼ��غͽ����طǺ�����ϳ�������һ����Ҫ����Ȼ��Դ���壬�����ڼ�����������������DZ�����ģ��������ù�غͻ��߾��ء��������ϳ��Ļ��ʲ�ͬ���Ǻ�����ϳ��ĵĺϳ��ɱ���Ǻ������ĺϳ�ø�Ļ���ؿ��ƣ���������Դ�Է�����ƣ��Ӷ������ṹ���ܶ������ģ���ʹ��Щ�����ܹ��˷���ͨ��ҩ��Ĺ��о����ԡ���Һ�Ͷ��ر���Ϊ�Ǽ�����������ĵı�����Ȼ��Դ�Լ�������Ȼ��Դ���绷�ĺ�������Ҳ�õ����о������á�ø�ϳ������ںϳɶ��ģ�����ĺ����ģ�ø�ϳ����ѳɹ�Ӧ����ʳƷ���Ӽ���ҩ���ũ�û�ѧƷ�������������ѱ����֤����һ��������������ĵĻ��������������ڻ��߾��ص�����������DNA�����ܹ���������ȷ�����к�ͬ���Ե��ĺ͵����ʡ����ַ�������������ж��������ij��Ļ������ر����ã�������Щ�ĺ���ͨ����ѧ�ϳɡ������ȵ��غ�����������ʹ������DNA������������������ҩ��Ĵ��������ӡ����⣬����DNA�����������Ŵ�������չ�������¼������ϣ�ͨ���������Ȼ�����Ὣ����Ĺ��ܻ�����������У�������������ϳ��ṩ��һ�����ķ�����ͨ�����Ӻϳ��ĺ�����DNA�����������������������Զ��ģ�����Ҫ���ж���˹���ʱ������һ���ر����õķ�����
ͨ���Ŵ�������չ���ж�������
��Ȼ�������� 20 �ֵ��Ͱ�����ϳɣ����������ұ��صİ���������������˵����ʽṹ���ܵĶ����Ժ����ԡ��Ŵ�������չ�Ƕ�ʮ��ǰ������һ�ּ�����ּ�ڿ˷���һ���ƣ�ͼ8 �� ���Ŵ�������չ�����ڵ����ʷ�������н������»�ѧ���������Եķǵ��Ͱ����� ( ncAA ) λ�������Եز������������Ķ����У�Ҫʵ����һ����Ҫ�ĸ���ɲ��֣�1���������軯ѧ���������Ե� ncAA��2��ָ�� ncAA �Ķ��������ӣ�����������ֹ������ (UAG) �������������ӣ�3�����ƶ����������Ҳ�������Դ��Ӧ������ŵ����� tRNA��4�����Խ� ncAA �����Եس�絽���� tRNA ���Ҳ�����Դ�������� tRNA �ϳ�ø/tRNA ��������������tRNA�ϳ�ø�������š�
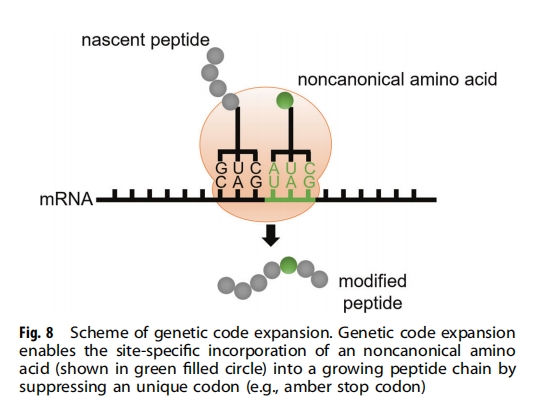
����Ϊֹ������ 200 ���־��в�ͬ���ܵ� ncAA ��������뵽��ͬ���������У�����˾�����ĸ�����鶯��ϸ���������������Ϊ�������о������ṩ�˱���Ĺ����䡣������չ�Ĺ���ģ���������������ѧ��ϰ��¡��������ϼ����⽻�������ڽ��������������ΰ����ᡢ������������εİ����ᣨ���ữ�����ữ�������ȣ���������ԭ���������Լ����⡢�˴Ź���ӫ��̽�룬�ѹ㷺Ӧ���ڵ����ʵ��о��������ͽ������Ŵ�������� ncAA ���������������Ż�������������ȷ�ṹ�����ܺͻ�ѧ�����Ļ�ѧ�������鵰�ס�����������ص��ע�Ŵ�������չ���������ĺ͵����ʽ����е�Ӧ�á�
�ĺ͵����ʵľ��Ҷ�����
ͨ���Ŵ�������չ�����Ķ̵��������Ƽ�������ҩ������ѧ�ϲ��������Ѫ�彵��Ϳ�������������˥��Ҳ�϶̡����Ӿۺ������ӳ��������Ƽ���˥�ڵ�һ�ַ�����PEG ���ظ��Ļ������鵥Ԫ�γɣ���һ�ֲ������オ�⡢����������ԭ�Եľۺ��PEG ���������ӵ����ʵ���Ч���������Ӷ�����������˵���������ʡ�PEG ���ֻ�����ͨ�����ӿռ�λ�������������ʲ�������ˮ��ø��������ͨ������Ŀ�군���ʵ�ˮ�����������������գ���Щ����ʹ�þ��Ҷ�������Ϊ���������Ե����ʵ��ձ���ԣ��� 1970 ������������Ҷ�������Ӧ�����Ż����������ƣ���ȡ���˾�ɹ���Ŀǰ�г����� 10 ���־��Ҷ��������������Ƽ������и���DZ�ں�ѡҩ�������ٴ������С�
��ͳ�ľ��Ҷ�����ͨ�����������������װ���л���Ȼ�������Ŀ�군���ʰ��������Ӧ�����������װ���л�������ȱ��ѡ���ԣ���Ͽ��������������Щ�л��е��κ�һ���ϣ��Ӷ��������Է�������ʽ�ϲ�������Ҫ����λ�������Ծ��Ҷ������ļ��������о��Ҷ������ֿ���ѡ���Եغ�λ�ÿ��Ƶ����ӵ��������ϣ�ͼ9����
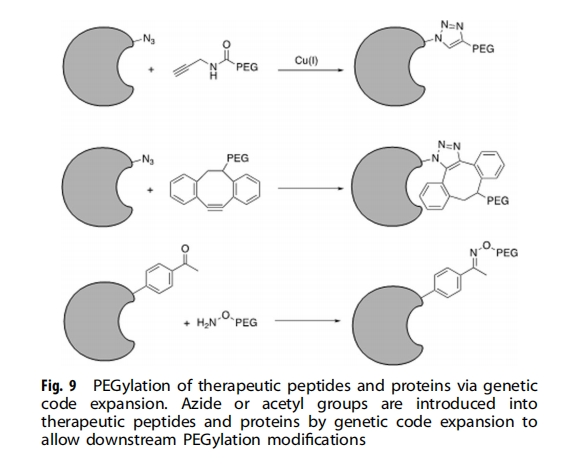
�Ŵ�������չΪ�����ʾ��Ҷ������ṩ�˱���Ĺ��ߡ�һ�ַ����ǽ���������������ѧ�ֱ��� ncAA��ͼ10���Ŵ����뵽Ŀ�군�����У�Ȼ��ͨ������������Ӧ�� PEG ��ϡ�2004 �꣬Deiters ���˱����˵�һ�ֻ��ڽ�ĸ�жԵ����������� (pAzF) ���Ŵ����ϵ� ncAA �鵼�ĵ����Ҷ�����������ͨ��ͭ (I) ����Ȳ��-�������ﻷ�ӳ� (CuAAC) �����Ӧ��Ȳ�������� PEG ��λ�������Ե����ӵ����������绯ø (SOD) �ϣ����� SOD ��ʾ����Ұ���͵��������Ƶ�ø���ԡ��ŵ��˱������ֵ����Ҷ���������Ҳ����Ӧ���ڸ����� (IFN)-��2b�������е�������� ncAA NEAK λ�������Ե����ϵ� IFN-��2b �IJ�ͬλ�ã��Ӷ�ͨ����ͭ���ӳɷ�Ӧ�� PEG ���������ͻ�ѧ������ϡ������ݶ���ģ���У��������� IFN-��2b ������ʾ�������������Ұ���ͷ��Ӹ��ߵ�������Ժ��õ�ҩ������ѧ�������������ӱ����������Ҷ����������ձ������ڸ��ֵ����ʡ�
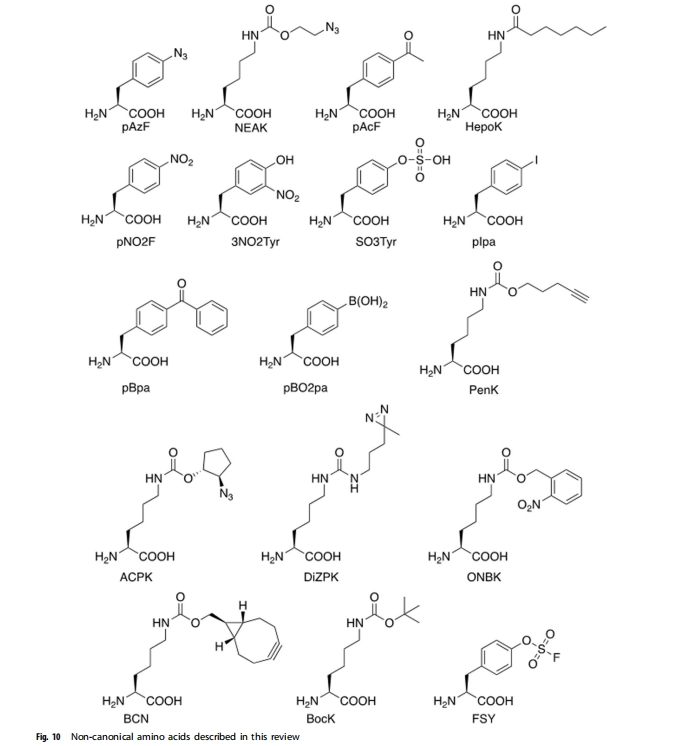
2011 �꣬Cho ���˱�����ͨ������������չ���������鵰���״��ٴ����顣ͨ���ڲ�ͬλ�ü���������������� (pAcF)��Ȼ���� PEG ����λ�������Խ�ϣ��Ƴ��� 20 �������������� (hGH) ���塣һ���ڲл� 35 �����е����Ҷ������� hGH ���壬��Ϊ ARX201���� GH ȱ���Ĵ����б��ֳ����õ�ҩЧѧ���ԣ������ڳ��� GH ȱ�������о�������Ȼ hGH �Ʒ��൱����Ч�Ͱ�ȫ�ԣ���Ч����ǿ��ע��Ƶ�ʽ��͡�Wu �������ͨ���� 35��131 �� 145 λ�㴦�Զ�� NEAK �����Ŵ����룬�������ڲл� 35��131 �� 145 ��������� PEG ���� hGH ���塣�뵥 PEG �� hGH ��ȣ����õĶ� PEG �� hGH ������ֳ����͵�����ԭ�Ժ��õ�ҩ������ѧ���ԣ��Ҳ���ʧ������ԣ����������ݶ���ģ���бȵ� PEG �� hGH ���и��ߵ��ȶ��ԡ���Щʾ��˵�����Ŵ�������չ�����Ż������Ե����ʺ��ĵ�ʵ���ԡ�
��һ��λ�������Ծ��Ҷ�����������ͨ���Ŵ�������չ���� PEG �� ncAA ֱ������е��ס�Shozen ����ʹ����ϸ������ϵͳ���������������ӣ�λ�������Եز����˺��� PEG4��PEG8 �� PEG12 ���� ncAA��Tada ����ʹ�����ƵIJ��ԣ�ͨ������������ֹ�����ӣ��� PEG4 �� PEG24 �Ľϳ� PEG ��������ġ�Fu ��������� eN-����-l-������ (HepoK) ���� GLP-1��ͼ10��), ����GLP-1(HepoK)��Ұ����GLP-1���, ����Ѫ�����(HSA)�Ľ��������ǿ, �ҽ�Ѫ�����ó���ʱ�����, Ϊ�о�������֬���ṩ�������Ĺ��ߡ�
�Ŵ�������չ�� ncAA Ҳ���������ɲ�ͬ���͵����磬���������磬���ǵ�λ���硢�������ͼ��������硣Grunewald�����״�֤��������ԭ�� ncAA ���ϵ�Ŀ�군���п��Դ��������������������Բ��ڶ���ģ�����������߷�Ӧ��������ԣ����������������� (pNO2F) ������ĵ�һͻ������С��������������-�� (mTNF-��) �ĵ� 86 λ���ֱ���� mTNF-�� (pNO2F) �� mTNF-�� (Phe)�����õ� mTNF-�� (pNO2F) ��С�����յ��ߵζȿ��巴Ӧ���� mTNF-�� (Phe) �ᡣ���⣬���� mTNF-�� (pNO2F) �յ��Ŀ�������Ȼ mTNF-�� �߶Ƚ��淴Ӧ��������С������֬���� (LPS) �յ��������������Ļ����о��У�Grunewald ���˷��� mTNF-�� (pNO2F) ͻ����ɲ��� T ϸ�������Զ��¡����Ϳ� mTNF-�� IgG ���巴Ӧ�����ַ�Ӧ���ٳ��� 40 �ܣ�������С������ LPS �̼�����������ڶ�Ѫ֢�����ַ����������˶����ӻƴ���ϵ��ĸߵζ� IgG ���巴Ӧ�������������ǽ�����������ԭ����������ת��Ϊ������ձ����÷������ں���ʵ��� ncAA��pNO2F���⣬�����������ض�λ�ò�����ϸ��ͻ�䣨mTNF-�� �е� Tyr �� EGF �е� Phe���ͷ�������Σ�3NO2Tyr �� SO3Tyr��Ҳ�����������Ȼ����ǿ�� IgG ���巴Ӧ���������������λ�������Բ�������ԭ�� ncAA ��ijЩ��Ȼ��������� (PTM) ���Դ��������������������Բ��������������硣
Wang ���˽�������б�������Ǽܵ� ncAA ���ϵ���˷�֦�˾���������ͽ�˷�֦�˾��У��Դٽ���˲�������о��Ϳ��������ڲ��鶯�ﲡ���Ĵ����Ժ��ӵ���װ���̣�ʹ�ó��滯ѧ���η������Ѳ��������Ļ����Ϊ�˿˷���һ��ս��Lin ���˱����˵�һ�����ӣ��� ncAAs λ�������Եز��������ͻ���У�Ȼ�����ѡ���Ա�ǣ�������ɥʧ��Ⱦ�ԡ�������˵��һ�����������������ﱻ�Ŵ����뵽�����ײ��� (HBV) �İ�Ĥ�����У������˸�ϸ������װ�ɻ���ײ��� (HDV)�������ϸ��ѡ���Ժ�Ч�ʡ�ͨ��ɸѡ��ͬ�IJ���λ�㣬�����Ĵ�Ⱦ�Ե�����ȫ���֡����⣬ncAA ���εIJ�������ͨ��ͭ (I) ����Ȳ��-�������ﻷ�ӳɵ����Ӧ���ɵر����»��ϡ�Wang ���˻����� ncAA �鵼�Ļ��ؿ����˼����� HIV-1 ���硣һ�鱽�����������ﱻ������뵽 HIV-1 �ı��走�������Կ����临�ƣ�ͨ�����ַ������Ծ�ȷ�شر� HIV-1 ���ơ��ں����о��У�Ԭ���˽� ncAA �鵼�Ļ��غϲ��������������У��������˻����������ƵĶ����ڿɸ��� HIV-1�������� HIV-1 ���翪������Ҫһ�����µ���ͨ��������Ȼ�����ʷ���ϵͳ��ʹ�õ����������ӣ�ʵ���˶� HIV-1 ���Ƶľ�ȷ���ƣ��Ӷ�����ȵؼ�����У�ԵĿ����Բ����������İ�ȫ�ԡ��÷�����Ӧ���ڼ������в������������˰�ȫ��Ч�ļ��������磬��Щ�����ڶ���ģ����������ǿ��ı��������߷�Ӧ������� ncAA �鵼�ļ�����������һ���ձ����õķ�����
������/������ҩ��
С���ӹ���ҩ����ǹ���ҩ����Ⱦ����������ƣ���������Ч�ʺ�Ч����ߡ�ҩ������ѧ���ơ�����ʱ���ӳ���������ҩƵ�ʼ����Լ��������е���ǿЧ�������á�������ѡ���Եͺ���ҩ��-�����ʼӺ����DZ������ԭ�ԵȰ�ȫ���⣬������������С���ӹ���ҩ�Ȼ�������ڻ��Եĵ����ʷ����������¼����ķ�չ��ζ��С���ӹ���ҩ�������ܵ���ע������ͨ�����۽�ϻ��������õ�С����ҩ���ѻ������С�
�������Ͻ������۵���ҩ��Ӧ�þ�����С����ҩ�����Ƶ����ơ�Ȼ�����������ǹ��е����γ���Ȼ�����ʵĹ��ۼ�����˹��۵���ҩ�������DZ����δ�õ���ֵ�̽����Li�������������һ�ֽӽ����Ӧ�Ʒ�(PERx)�������������۵���ҩ����ǽ�DZ�ڵ��������ncAA������- L -�Ұ���(FSY)268�Ŵ��ؽ�ϵ��˳�����ϸ����������1(PD-1)��129λ�����������õ���PD-1(FSY)�����������ѡ���Ե�������Ȼ����PD-L1�γɹ��ۼ�������עĿ���ǣ���Ұ����PD-1��ȣ�PD-1(FSY)������ǿ����naïve Tϸ������Ƕ�Ͽ�ԭ����Tϸ����������ԡ��ڼ���������Դ��С��ģ���У�PD-1(FSY)��ʾ������Ч�����������������ã��������������Կ�PD-L1�����൱�����Ŀ��������á�Ȼ�����ǽ�PERxӦ����fsy���ε�ճ�����HER2����Ĺ������ƣ�˵��PERx����Ϊ�������۵���ҩ���ṩһ��ͨ��ƽ̨����ǹ��۵���ҩ����ȣ�PERxҩ���������ԭʼ��ʽʹ�ã�����Ҫ������������ӳ����˥�ڣ���Ϊ���۽��ʹҩ��ҩЧ����ҩ������ѧ������⣬PERx����PD-1 (15.6 kDa)��С���������Ƽ���������ҩ��Ӷ������չ�����Ƶ��ķ�Χ�����⣬���ڵ�����ҩ������б�֮��������������Լ�DZ���������ncAA���ڽ������������ƣ�PERx��������ȵؼ����Ѱ�ЧӦ����Щ������ζ��PERx������DZ��Ϊ���������۵���ҩ���ṩһ��ͨ��ƽ̨��PERx���Ա���Ļ�ѧ����Ĺ��۵��������Ѿ��������ط���ϸ�ع�.
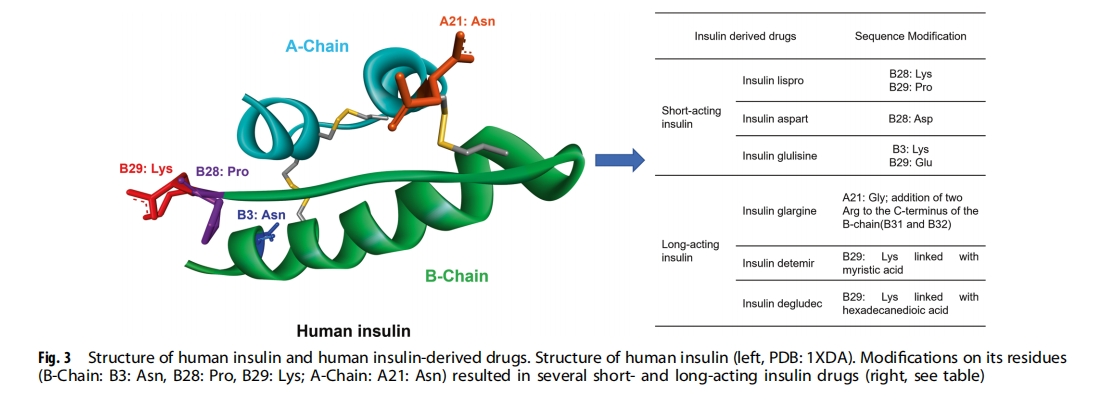
֬�ʺͽϴ�ĵ����ʾ�����ż���Ը��ƹ�����ҩ���ҩ������ѧ���ѻ�������ҩ�������³�ġ�����³�ĺ͵¹��ȵ��أ��� C 14/16/18 ֬�����ϣ������������ǵ�Ѫ��ѭ��ʱ�䲢�������������������������еĽ��⡣����Ѫ�����ף�Ѫ����������ף�Ҳ����ͨ����������������ӳ���ѭ��ʱ�䣬�Ӷ�������С���˹��ķ�������ֵֹ�����磬���ֲ��Ա������ӳ�����³�ĺͰ���³�ĵİ�˥�ڣ�ÿ��ע��һ�Ρ�
��ҩ�����͵ķ�չ
�����ο�ʹ�Ļ�ø��õĻ��Ժ�Ѫ���ȶ��ԣ�����ø���ҩ�Ȼ�����ĵĹ���������ζ�����Ǻ����ױ�θ�����е�����øˮ�⣬��˴��������ҩ����ͨ��ע���ҩ�ġ�������о��Ѿ�̽��������ҩ��ĸ�ҩ;���Կ˷���Щȱ�㡣
�����ٽ�������ʹ����һ�ֺ���ǰ���IJ��ԣ���ʵ������ҩ��Ŀڷ���ҩ���� C18֬�����ϵ�����³���ѻ�ͨ��ÿ��һ��Ƥ��ע���ҩ, ��Ѫ���ȶ�����������GLP-1����������˹�����ǣ�����³����N-[8-(2-�ǻ�����������]������ (SNAC) �ĸ����Ƽ��ѻ������ڿڷ����� 2 �������� SNAC �ĸ����Ƽ��ɽ�������ø�Ĺ�Ч���Ӷ���ֹ����³����θ�б��ƻ�����ˮ�� SNAC ���ӻ�����������³�ĵ���֬�ԣ��Ӷ�������ͨ��θĤ�Ŀ�ϸ�����ռ��������ѭ����ת�ˡ����������ٽ�����ø���Ƽ���ˮ�����ĸ����Ƽ�Ҳ�����������ڷ���������ҩ�������ĺ��ȵ��أ���Щҩ��Ŀǰ�����ٴ������С�Ŀǰ�����о�������ԣ������β���ҩ��Ƥ��ҩ��ʹ��ֲ��ʽ�ã����������ض�����ҩ�������������ʽ�ȵ��غ������ȵ������͵���ֲ��ʽ�á�����Ԥ����Щ��������δ������Ӧ���ڸ�������ҩ�
���������ڼ�������ķ�չ��Ӧ����״
�������������������е�Ӧ��
2 ���������ɺ����ȵ���ȱ������ģ������������кܳ�����2 �������ѳɹ�ͨ������ҩ�����ƣ����� GLP-1 ���弤���� (GLP-1RA) ��������������ҩ���ȵ��ء�GLP-1 ��һ���ɻس� L ϸ�����ڵ���Դ���������ء���������������� �� ϸ�������ܺ�������ϵͳ�������Ѫ�ܡ����ࡢ�κ�θ����ճĤ�У�ͼ11����GLP-1 ������������ã��̼��ȵ� �� ϸ�������ȵ��أ������ȵ� �� ϸ���ͷ��ȸ�Ѫ���أ����ӱ����У����������������ķ�ʽ�ӻ�θ�ſա���Դ��GLP-1�����Ļ���ø-4��DPP-4�����ⲢѸ��ʧ�Ϊ���ӳ�GLP-1����Ĵ̼�ʱ�䣬��ϳ�GLP-1RAs����ֹ�併�⡣��2005���һ��GLP-1RA�����������ı�����ʳƷҩƷ�ල�����֣�FDA�������������GLP-1RAs�����ٴ�����������³�ģ�2009�꣩�����������ģ�2013�꣩������³�ģ�2014�꣩������³�ģ�2017�꣩����ЩGLP-1RAsע�������Ч�����ǻ�Ѫ�쵰��ƽ��Ѫ��ˮƽ�����ƿո�Ѫ�ǡ�
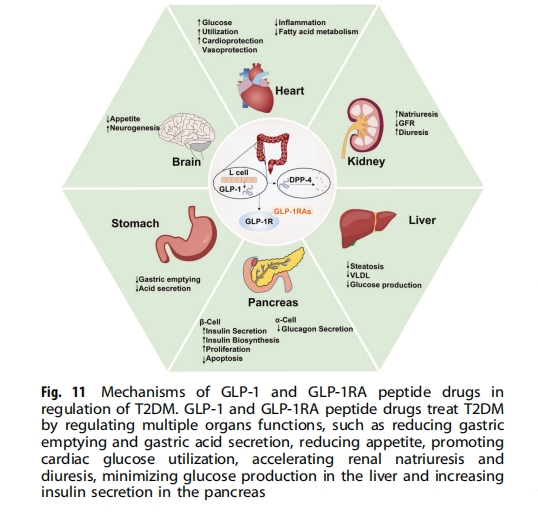
һЩGLP-1RAs������ijЩ2��������֢Ҳ��Ч������������2��������Σ�յIJ���֢֮һ���������ߵ��������������Ӱ�죬�ٴ����ְ������������С���˹��ʣ�GFR���½�����һ���35��2�������ߵ��о��У�����³��ͨ�����ƽ���С�����ⷴ��ת�˵���3��NHE3���Ӷ������ơ��Ⱥͼصľ��ԺͲ�����й����������ҺpHֵ���Ӷ�������þ���ƺ������εľ��ԺͲ�����й����ʾ��ȵ�����ȡ����⣬��һ���30��2�������ߵ��о��У�����³������������GFR���������й�ʺͲ��ְ�����й . GLP-1RAs ��ͨ�������������ܰߵ�������������С��-��С�����ʹ���С������Ѫ������������ GFR�����һ�����ͨ������Ѫ�����ػ��ԡ�������������Ӧ�����������������ٰ�����Ȼ������ЩӰ���ڶ��̶������� GLP-1R �鵼�����д�ȷ����������о�֤ʵ��GLP-1 �Ĵ�л�ﱣ������Ҫ�Ŀ������Ϳ��������ԣ���Щ������ GLP-1R �ء���Ѫ�ܼ�������2����������������Ҫԭ�������ѡ��2���������Ʒ���ʱӦ������Ѫ�ܲ���֢�ķ��Ρ�GLP-1RAs�ѱ�֤ʵ����Ѫ�ܼ������������á�������ٴ����鷢�֣�ֻ������³�ĺ�����³������Ѫ���洦���������ƣ�����������в���ȷ�����ܾ��п���������Ӳ�������á�����GLP-1RAs����Ѫ�ܼ����ı������ò����ԣ�����������ȫ����û���к�Ӱ�죬���GLP-1RAs�ķ���-Ч��ֲ��Ǿ���ġ�GLP-1RAs �� 2 �������ߵķ���֢״Ҳ���ֳ��������á�Thondam ������һ���������������Է��ֺ��ַ�����ز���֢������ 2 �������Ļ��߶������ķ�Ӧ���ã����غ�Ѫ�ǿ������Ը��ƣ�������ͨ�����ӱ����кͼ������������������ڻ���ʵ�ֵġ�һ��� 25 ������ 2 �������ߵ��о����������ܶ���˫�Һͻ�������/DPP-4 ���Ƽ����� 6 ���µĻ��ߣ����ͬʱ���� GLP-1RA���������� 19��6 ������ƽ�����ء��ǻ�Ѫ�쵰����֬�ʾ���������. 25 �����ܰ������ĺ�����³������ 3 ���µ� 25 �� 2 �������ߵ�����ָ����֬�����Ҳ�����½�. 2 �����ɵ��¹��ʴ�����ӹ��ۺ������ϲ����ȹ���ز���֢�ķ��ա�ʵ���о����֣�GLP-1RAs �Թ��ʺ�ǿ���������Ļ������ã�������ͨ�����ƹ�������������Ĺ���ѪҺ��Ӧ��һ���о�������³��Ӧ�����ѳ��г��� 2 �����������Թ�����Դ��������С RNA (miRNA) ���и�ͨ�����������������³�Ŀɵ��°����ȵ����ź�ͨ·�������� miRNA ���������仯��������������鵼�� Wnt/��-catenin �ź�ͨ·�ı仯��DZ��������йء�
GLP-1RA��������ĸ�������θ������ز�����Ӧ������ġ�Ż�º�к����ע�䲿λ��Ӧ������ЧGLP-1RA�����ý��٣���ҩƵ�ʽϵͣ������Ը��á�����˫�������ٴ�����2������һ��ҩ�����ŷ�������о�Э�����������Э��Ľ��飬���ڵ��ö���˫��Ѫ�ǿ��Ʋ��ѵĻ��ߣ�����GLP-1RA���������ࡢ�������ͪ�ࡢDPP-4���Ƽ�����-������Эͬת�˵���2���Ƽ����ȵ�����Ϊ����ҩ�������GLP-1RAs���˿���Ѫ��֮�����������洦�������������ࡢ������Ѫ�ܼ������ա��������ء���Ѫ�Ƿ��ա��Թ���֢״���桢�����÷����ʵ͵ȣ�GLP-1RAs��δ����T2DM�����бؽ����Ӳ�����������á�
������������Ѫ�ܼ��������е�Ӧ��
�ڷǴ�Ⱦ�Լ����У���Ѫ�ܼ�����Ŀǰȫ��Χ�ڵ��������ͷ�������ߵļ�������Ѫѹ����Ѫ�ܼ�������ҪΣ������֮һ������Ϊ����������-Ѫ�ܽ�����-ȩ��ͪϵͳ��RAAS���ͽ�����ϵͳ���Թ����Լ����������¡�RAAS�е�Ѫ�ܽ�����ת��ø��ACE���Ĺ����ǽ�Ѫ�ܽ�����I�ѽ�ΪѪ�ܽ�����II��ʹѪ���������������Ѫѹ����ACE2��Ѫ�ܽ�����IIˮ��ΪѪ�����ż�Ѫ�ܽ����أ�1-7������ӽ�Ѫѹ����ˣ����RAAS�ǿ�����Ѫ�ܼ�����������ԡ��ϳɵ�Ѫ�ܽ�����II��2017����FDA��������ͨ��������ע���Ӱ�Ѫ֢�������ֲ�ʽ�ݿ˳��˻��ߵ�Ѫѹ��Montone���˴�б�����������з���ɸѡ���������ģ�WPRGYFL��GPDRPKFLGPF��WYGPDRPKFL��SDWDRF����ʾ����ACE�����ƻ��ԡ�Liao���˷������Ե��������IRWͨ���ϵ�ACE2�ı����������Է��Ը�Ѫѹ�����Ѫѹ����Щ�о�������RAASΪ�е��ʳ����Դ����������Ѫ�ܼ����������DZ�ڵ�Ӧ�ü�ֵ��
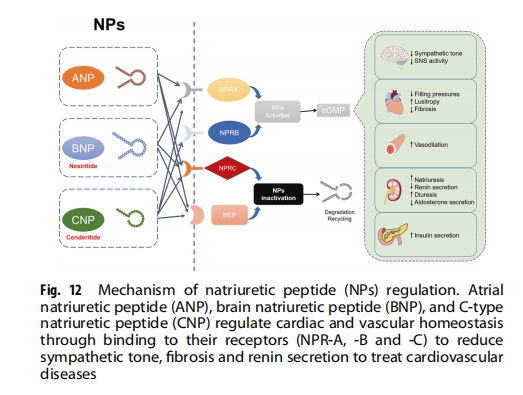
������ (NPs) �������ķ������� (ANP)���������� (BNP) �� C �������� (CNP)���������Ѫ����̬����Ҫ���ڼ���ͼ12������ˣ���� NPs ��Ԥ����������Ѫ�ܼ�������һ��ʵ�ò��ԡ�����������һ�������� BNP��2001 �걻 FDA ���������ƾ�Ϣ����Ⱥ������ѻ��ߵļ���ʧ����������˥�ߣ�Ȼ���������������ԵͺͰ�ȫ�����ޣ�����δ�õ��㷺Ӧ�á�NPs ��Ҫͨ�� NPR-A ��/�� NPR-B ���������ã��� NPR-C ��Ҫ������� NPs��Cenderitide ��һ��˫�� NPR-A/NPR-B ���������� CNP �ʹ����������з�������۾��������ĵ� C ����ɡ�Cenderitide Ŀǰ�������ٴ��о��У�������ʾ����������˥�ߺ���˥�ߵİ�ȫ�Ժ�DZ�������⣬һЩ����Ѫ�ܼ�������������ڶ������Ͻ��в��ԡ����磬�ڴ�������עѪ�ܻ��Գ��Ŀ������ļ�Ѫ�ܻ��Գ��ĵ�Ũ�Ȳ���ת���е��ļ���ά�������� RD808 ���к� �� 1 -�������������壬�Ӷ����� �� 1-�����������塣�����������Ƥ�ʼ����ͷ����� (CRF) �����ϵͳĿǰ��ΪԤ����Ѫ�ܼ����İе��ܵ�Խ��Խ��Ĺ�ע��CRF�����ϵͳ����Ѫ��ϵͳ֮����ڸ��ӵĹ�ϵ����������Ѫ�ܹ����е�ȷ�е����������д�ȷ�������⣬�� 2 ���������У�ѭ�� DPP-4 �������ӣ�Ѫ���鵼���Ž��͡�Ѫ���鵼�����ǹ��ϵ���Ƥ�����ϰ��������־�Ҳ��δ����Ѫ���¼���Ԥ�����ӣ������ DPP-4 ������Ԥ����Ѫ�ܼ�����DZ�ڰе㡣
����������θ�������������е�Ӧ��
���������ڳ������������е�Ӧ��
�������У�θ������Ⱥ���ɸ��ӵ���̬ϵͳ��ͨ��������θ������Ⱥ���ɸ��ӵ���̬ϵͳ��ͨ������Ƥͨ���ṩ�������Ϻͷ��ڸ��ֿ������ӣ����������ģ�AMPs�������ڳ��Ĥ���泦����Ⱥ����ɡ����ƾ�Ⱥ��������Ⱥ��������Ⱥ���²��������ֶ�̬ƽ�⣬������ϸ���������������桢ʳ���ж���ҩ�ﲻ����Ӧ���Ŵ����ص�����ĸ��ֳ��������У��糦�ס����ء���������֢�Գ�����IBD���ȣ��������Ķ����Ծ����ƻ��������صĴ���ʹ�ÿ��ܽ�һ��������������������ԣ����������ƣ��������ܼ��ز��飬����IBD������ȷ��ǰ2-5���ڸ��п���ʹ�ù������ء�����ҩ���������ԡ���Ч�ԡ��Ͷ��Ե��ص㣬�ڸ����������˹㷺��ע��
����������Ⱥ�������ı������-���ﹲ����ϵ���ƻ������� IBD ������չ�Ĺؼ���IBD �����������������Խ᳦�ף����ɳ������߷�Ӧ����ģ������֢���ɻ������Ŵ��������������ġ��� IBD �ľ��巢�������в���ȷ��Ŀǰ������Ч�����Ʒ�����IBD ���߳���������������Խ��ͣ����������ź�ھ��ţ�ë�ݾ��ƣ�����˾������Լ��٣������θ˾�����������. ����֤�ݱ������θ˾��ų�Ա�� IBD�з��ӹؼ����á�������-������-39 ��һ������������ܰ���֯��Ȼ���ڵ�С������ AMP����֤ʵ���п��������ߵ��ںͳ���Ƥ�����ܣ�����Ϊ IBD�ṩ��ȫ������Ʒ���
����������ͨ�����ܳ��г������ƣ����¶̳��ۺ�����SBS����С�����˺ͳ���ʱС���쳣��СҲ���ܵ��� SBS���䶨��Ϊ�����С�����ȳ���С�� 200 cm ��ص�֢״��GLP-2 �ɳ��ڷ��� L ϸ����������ϵͳ�ĸ�����Ԫ������ͼ13������������GLP-2��SBS�������ܵ��㷺��ע��GLP-2�ѱ�֤ʵ���ж����������ã������̼�������ϸ�����������ٳ�ϸ���������ٽ����Ĥ���š�����θ����ں�θ�ſա��̼�����Ѫ������ǿ�������Ϲ��ܡ�������֢���ˡ��ٽ�Ӫ����Һ�����ա�GLP - 2�����ڰ�����ת�˵��ı��ֱ�Ӽ���mTORC1�����ӳ�����Ƥ����������ա�һЩ�ض��İ����ᣨ�����Ȱ��������Ȱ��ᡢ�����ᡢ�ʰ��ᡢ�����ᡢ������ͺ����ᣩҲ��֤����ά�ֳ��������Է��淢����Ҫ���ã�������ֹ����ή�������Ƴ������Ϲ��ܡ�������֢��ϸ����������Դ��GLP-2�ױ�DPP-4���⣬��GLP-2���������³��ͨ����GLP-2 N�˵ڶ�λ�ı������滻Ϊ�ʰ��ᣬ����˥�ڴ�7�����ӳ���Լ2-3Сʱ����Ч��ֹ�䱻DPP-4���⡣�ٴ��о����������³�Ŀ���Ч���ٻ������Գ���Ӫ����/������Һ֧�ֵ���Ҫ�������³����Զ�˻س��г���������Ӧ�����������Ӳг���λ�����͵����ʺϳɡ����³����2012�걻FDA������SBS���ߵ��ٴ����ơ�Wiśniewski���������һϵ��GLP-2���������2-�ʰ���ȡ����10-��������ȡ����11-��/��16-��ˮȡ���ȣ����������GLP-2R����Ч������Ȼ���أ����ֳ����õ�����ѡ���Ժ͵�ϵͳ����ʡ����У��ĶΣ�([2Gly, 10Nle, 11DPhe, 16Leu] hGLP-2-(1−33)-NH2 ����ѡΪ�ٴ�������ѡҩ��. �ȸ�Ѫ����ԭ�����е� GLP-1 ������ GLP-2 ���ƵĹ��ܣ����ѱ������������� SBS����һ���о��У����� SBS �����ڽ��� GLP-1 �����������������ƺ��ű�Ƶ�ʺ���̬���������ơ�ͬ����GLP-1 �����˾��� SBS ���ߵĸ�к����Ч������ GLP-2���� GLP-1 �� GLP-2 ��������ڵ���ʹ�������κ�һ�֡��ȸ�Ѫ����ԭ�������һ����Ա�����ȵ����ƺ�Ҳ������������̣����糦��Ӫ�����˶���θ����ڣ���������������ȵ������ĵ�ǰ��. �������������� EGF���ٺ�ϸ�������غ�ϸ����������Ҳ����ʾ���� SBS ������DZ����EGF �� GLP-2 ��������������� SBS ������ģ�͵�С�����ȣ����� EGF �������������� SBS ��DZ�����ٺ�ϸ��������ͨ���̼�����ģ���н������ӵ��ı����������������Ϲ��ܲ�����θ��������ȱѪ/�ٹ�ע���ˣ�����ע���ϸ���������ӿɽ��ʹ�������С���᳦�ķ����ʺ����س̶ȡ�
��������²���������ļ����������A�������Ⱦ�ߵĸ�к����֢���������ص�αĤ�Խ᳦�ס������-2��YPCKLNLKLGKVPFH����Ji�ȴ�������з���õ���AMP������ϼ����������A������Ĥ���˺���֢ ���ǽ��ڱ��϶�Ϊ����/���Ƽ����������A�����αĤ�Խ᳦�ĺ�ѡҩ��. �Ӻ��������з������ 9 ���������������� CopA3 (LLCIALRKK) ���������˼����������A �����С����֢��Ӧ�����Գ��ף�������ȫ�����С���Ͼ�����������������Խ᳦����֢��Ӧ������������Ӧ ��������Ĥ���A �������ʳ���ж��뼸����Ҫ������θ���������йأ�����Ϊ���ɲ�����Ĥ����IJ����鵼�ij����� (CPE) ���˳����������ӵ���ϡ�Archana ���˷��֣��� CPE ��������ӵ���-4 ��� ECL-2 ��Ԥ������������������ CPE �յ����ó���344ǻ��Һ����ۺ���֯ѧ���ˣ�������ϳ��� ECL-2 ���ܴ���һ��Ԥ��A �Ͳ�����Ĥ�������ij�����֯ѧ���˵�DZ�ڲ��ԡ��˽᳦��Ƥ���ڵĿ���������һ�־��й㷺���������ߵ��ڹ��ܵ� AMP��������о��������˿��������������ڽ᳦��Ƥϸ��������Դ�����˺�ɳ���Ͼ���ֹϸ�����֡�ά����Ƥ���ϵ������ԣ�����ͨ������Toll������4�ʹ���ϸ��������ʵ�֡����⣬����������ȾҲ��֤���ܴ̼�AMPs�ı���µ��о�����С���Ǻ��Ს����Ⱦ���ӳ���Ƥϸ���˦·�����3�ı���ͷ��ڣ��˦·�����3����ϸ���⿹�����������ԡ�
������ά�� (CF) ����ͨ��������ֳ�����ͱ��أ����ܷ�չΪԶ�˳������ۺ����������ỷ��ø C (GCC) ���弤�������������� 2012 ��� FDA �������������Ա��ء��������Ļ���֤�����Ը��� CF ģ��С��ij������䣬���ܻ���Ҫ��һ���о���������� CF ���߳���������Ӱ�졣NHE3 ���Ƽ� tenapanor ͨ������������������ CF С���θ�������䣬��������� GCC �ź�ת���� NHE3���������� CF ���߱��صĺ��ʰе㡣
����ҩ���������⣬��Ҳ��������ʳ�Asn-Pro-Trp-Asp-Gln (NPWDQ) ��һ��ͨ��ˮ���ҵ��ף�һ����Ҫ��ţ�̵��ף���õ��ģ�������������ʳ�����ԭ���嵰���������峦�� Caco-2 ϸ���У�����������Ŀ��ܸ��Ƴ�����Ƥ���ϵĹ��ܡ���-Casofensin ��һ���ڷ������з��ֵ��ģ�����ʵ�鷢�֣����ڷ��� ��-Casofensin ��ͨ��������״ϸ���ʹٽ��˿�����������������������ij������˺���֢��������������ij������˾������������ͬ���ٴ�����֯ѧ�Ͳ�������ѧ������ʾ����-casofensin�����ǿ�������DZ�ڸ������Ʒ�����
����ҩ�������ݳ�������������Ҳ���Ź�����ǰ������������Ȼ�ߵ���Ϊԭ�ϣ����Ƴ������ϳ���KR-32��KR-32�ɸ��Ʋ������ش˾�K88��Ⱦ������֬�������ղ�����������ȡ���������ʼ�������̬����ʾKR-32����DZ�ڵ�ҩ�ü�ֵ��C-BF��һ����Դ�ڿ����ĵĶ��ģ���AMP��������Ϊͻ����һ��������Ϊ������ǰ;�Ŀ��������Ʒ��C-BF���������˶���������������������LPS��С���Ľṹ�ͷ������ˣ������C-BF����������LPS/��ԭ������ij������˵�DZ��ҩ�
����������θ�������е�Ӧ��
θ�Ĥ������Ͷ������������֮֯һ��θ���dz��������⡣�����ݸ˾���Ⱦ�������忹��ҩ���ƾ������̡�������ѹ�������θ���˵���Ҫ���أ���������θ������θ��������ʱ���ƻ����Ʋ������ᷢչ�����Բ��������ij������˻�������θ���ķ��ա�θ����Ŀǰȫ����Ĵ���֢�������Ժ�Ů��֢����ԭ���зֱ����������͵��塣
��ȻĿǰ��û������ҩ�ﱻ����������θ��������ʮ����������ҩ�������Դ�Ժ���Դ������ҩ����θ���е����õõ��˹㷺���о��������ػ�������ģ�CGRP���㷺�ֲ���θ����ϵͳ�������������ио�����Ҫ���ʡ���Щ�о����뱣��θճĤ���ܸ��ִ̼���CGRP�ڴ˹�������DZ�ڽ������ã�����θճĤѪ��������θ����ڣ���ֹϸ���������������ˡ���CGRP�⣬һ��������ø-һ��������NOS-NO���ͻ�����ø-ǰ�����أ�COX-PG��ϵͳ��θҲ�����Ƶı������á�CGRP��NO �� PG ����Ϊ��θ�������ն˽��ʣ����鵼������Դ���ĵ�θ�������á��Ҵ������θ���˵���Ҫ����������θѪ�����ˡ���Ϊ�����������յ� (VGF) �����������ģ�TLQP-21 ������� NO��PGE2 ���������ؽ鵼�����������������ע������Լ��������Է�ʽ�����Ҵ������θ����, Novokinin (Arg-Pro-Leu-Lys-Pro-Trp) �����Ѽ������ε���ЧѪ�����źͿ���Ѫѹ�ģ���Ѫ�ܽ����� II 2 �� (AT2) �����нϸߵ�ѡ�����������ŵ��о����֣�novokinin �Լ��������Է�ʽ����������ע������θ����ڣ�����θ�Ĥ���ܾƾ���������ˣ�����ͨ���鵼 AT2 ����-PG ͨ·����Щ�����ʾ�� TLQP-21 �� novokinin ��θ���������еļ�ֵ���ӷ�ơ�ƽ�ĸ����ˮ���� (������ < 3 kDa) �л�õĶ�����ȡ��ɼ������θ�Ĥ���ˣ���ʾ��ĸ������ȡ����θ�������е�DZ�ڼ�ֵ��
����Ӧ����θ���˳��������о�Ӧ����θ�����Ƶ�ģ�ͣ�AMP�����ر���Ϊ���ɱ�ϸ������θ�����ɶ������ģ��ó������ص�С��θ��������Լ��٣���ʾ�����ؿ�����Ӧ��������θ����ķ����йء�nesfatin-1������ʳ�ļ��壬�����ڳ�������Ԫ���ڷ���ϸ���С�Alexandra�ȵ��о�������nesfatin-1��ˮ������Ӧ������θ�����Եı������ã��������θҺ���ڼ��١�COX-PG��NOS-NOϵͳ�鵼�ij�Ѫ���������о������ȩ����ļ����йء������º�Ӧ�����������θ����������������������Ŀ�ͨ������ϸ����������֢������������θ��.�����������ע��߲�����������������Ӧ������IJͺ�θ������ǿ���Ӷ�����θ�ſ��ӳ٣���ʾ�߲��ؿ���������Ӧ�����θ�������ϰ��ĺ�ѡҩ�
θ����һ�����ص�θ�����������ֶ�����θ����������ʾ�����õ�ǰ����GEBP11��ͨ���ɾ���չʾ����ɸѡ�ͼ�����һ���µľŰ�����鳲�ġ�GEBP11ѡ���Ե������꾲����Ƥϸ��������Ѫ�ܽ�ϣ���ʾ���������������Ͱ���ҩ����͵���Ҫ��ѡҩ���131��ǵ�˫��PEG��GEBP11�����壨131Imur2PEG- (GEBP11) 3�����ƿ���������������θ��������ֲ�����������ӳ�����ʱ�䣬��ʾ131Imur2PEG- (GEBP11) 3������θ���İ������Ƶĺ��ʺ�ѡ�Ҳ��θ����Ѫ���������Ƶ�ҩ�����塣�����ݸ˾���Ⱦ��θ������Ҫ����֮һ��HP-6��Pro-Gln-Pro-Lys-Val-Leu-Asp-Ser���Ǵ���������ˮ������з���Ļ����ģ��ѱ�֤�����еֿ������ݸ˾��յ����°��ԡ�HP-6ͨ������EGFR����µ����ἡ��3-��ø/Akt�ź�ת���ͦ�-catenin��תλ����Ч���������ݸ˾��յ�����θ�ٰ�ϸ����AGS����ֳ��Ǩ�ƣ���������ϸ��������AGSϸ����Ϯ��Zhang���˺ϳ���AMP pexiganan������������PNPs��������ֳ��������ݸ˾����ԣ����ұ�pexiganan��С��θ�е������ݸ˾��и�ǿ���������������ʾ�����ƺ�Ԥ�������ݸ˾����θ����DZ����TFF1��һ����𤵰����ص�θ�Ĥϸ�������ġ�TFF1�ı����������ݸ˾���Ⱦ�ļ����ڶ�����������θ����ϵ�С���Ⱦ����������֢��Ӧ�ʸ���أ����� TFF1 ����������ϸ���ֿ�ϸ����������֢�ķ�չ��ͬ��һ������� TFF2 �ѱ�֤������θ���� MUC6 ����ã��ȶ�θ�Һ���ϣ�ά��θ�Ĥ�����ԡ�
������ҩ����θ��������Ҳ�е������á�����ע��GLP-2ͨ������CGRP����Դ��PGs����NO������θ����Ѫ������θճĤѪ��������Դ��GLP-1������ȫθģ����ͨ��������NO�ͷŵ�θ�Ӷ�����С��θ���������ѻ�����GLP-1/2�������Ƿ���������������д��о���BNP��������Ѫ�ܵ����ԣ��������������ע�����ϣ�����BNP�ѱ�֤ʵ������θճĤѪ���е�Ѫ�쵰������. θ���غ��������ͷ�������ͬһ�ļ��壬��Щ������θ�����˶�������������Ҫ���á��������ͷ��ĺ�θ���ؿ��������������Эͬ�̼�ǿ�ҵ�θ������θ�����Լ�θ���غ��������ͷ��ĵĽ��ͨ���鰷�鵼��;���̼�����θ����ڡ�
���������ڰ�֢�����е�Ӧ��
��ͳ�İ�֢���Ʒ������������ͷ��ƣ������ڰ�֢������Ч���ޡ����������ƺ��������Ƶķ�չ���������˰�֢���ߵ������ʡ�����������������ϸ�����ض����ӻ��ź�ͨ·���������ԡ��������ķ�ʽɱ������ϸ������������ҩ�ﲢ��ֱ�ӹ�������ϸ�������ǵ��ڻ�������������ϵͳ��ͨ����������������������ϸ����PD-1/PD-L1��������֪�������㣬Ŀǰ����5�����PD-1/PD-L1����õĵ���¡���屻FDA���������ư�֢����������ڳɱ��ߡ��ڷ���Ӧ�Բ����ԭ��ǿ��ȱ�㡣�������ڷ��ӳߴ�С�������ߡ������Ρ�����ԭ�Ե͵��ŵ㣬��������Ϻ���������Ҳ���������ǵĹ�ע��һЩ���κ�Ķ��Ļ����ֳ����õ��ȶ��ԣ�����Carvajal���˿������ȶ��Ħ�-��������ΪMDM2��MDMX�����Ƽ�����������p53������֢��
��Ȼ�������ڵİ�˥�ڽ϶̣��������ϸ���и����쳣�����������ͨ���Ǿ������ε����������Щ�ĵ�������Ҫ�����ַ�����������ȱ�㣺1������Ȼ��������������2����ѧ�ϳɺͻ��ڽṹ�ĺ������̻���3���Ŀ�ɸѡ�����У��ɾ���չʾ������һ�ִ�ͳ��Ӧ�ù㷺�ķ��������в���������Чɸѡ������ͬ�ĵ��ŵ㡣
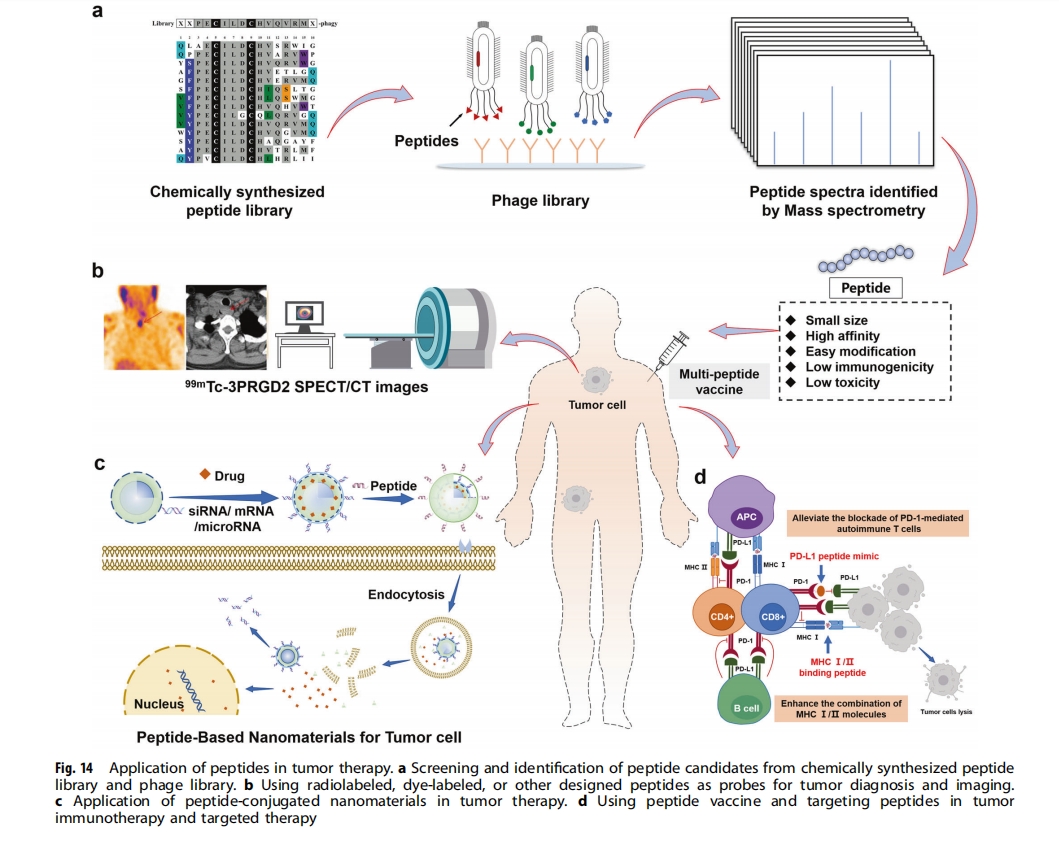
���������������е�Ӧ����Ҫ�����ַ�ʽ��1�����÷�����ͬλ�ء�Ⱦ�ϻ������ѱ����ķ��ӱ�ǵĶ�����Ϊ̽�룬����������Ϻͳ���2��������ż�������ײ��Ͻ����������ƣ�3�����������缤������ϵͳ����Ԥ����4������ʹ������Ϊ����ҩ�ͼ14����
�����ĵij���̽���������������Ա���������ϡ���Щ������Ա�����ϸ�����棬���� ��v��3 ������ (RGD ��)��EGF ���塢�����������塢��ѹ�������ת���������壻Ҳ���Ա�����ϸ���ڣ����� Bcr/Abl��ϸ�����ڵ��� A ��ϸ�����ڵ���ø��Ҳ���Ա�����ϸ��������У������������ס����ʽ�������ø��ǰ���������Կ�ԭ������ͨ�������ӷ��������ϲ�ɨ��/������ϲ�ɨ����ӻ�̽���λ����ָʾ�����ֲ����ü�����Ӧ��������������Ϻ������г����Ѿ������˼���̽�룬��������ĺ͵������ģ���Ϊ�����������ĵķ����Ա�ǽ����ѱ�FDA���������������������ڷ��������ͷΰ� �����ҵ��ǣ����������ѱ����ء�����RGD�ĵķ����Ա��������ܵ���ע�������Ѿ��ϳ���һϵ��̽�룬������99m��Tc-3PRGD2����������ͨ�������Ե�387������ȫ��ɨ����ֻ��ͼ�״�ٰ�. �� 177 dotatate ��һ�ַ����Ա�ǵ��������������������������������������������Ե�θ�������ڷ��������������������������ϣ�Ȼ���ͷŷ������� 177 ��������ϸ����ͨ���γ�ϸ�������ɻ��յ�ϸ�����ˡ�
�������Ļ�̽����ͬ��ԭ��������ͨ�������ϱ�Ǧ·�������ʵ���ڲ��������ƣ�Ȼ�����������ܵ��Ծ��������������������֯�ķ������˵����ƣ���Щ����λ������������Զ����һ�ּ���DZ�ڸ����õĿ�����������ǽ����뿹��ҩ������RNA��С����RNA��[siRNA]/miRNA/mRNA����ż�����͵�����AN-152��AN-207���밢ù��ż���Ĵٻ��弤���ͷż���������Դٻ��弤���ͷż����������Եİ�֢���п������ԡ�I�ں�II���ٴ��о������������ҩ�����ٰ����ӹ���Ĥ�����ѳ�����������Ч�����Ժ������еȡ�Chen ������˾��Ҷ�����֬����-��������-DNA (LPD) ��������ͨ�����ΰ����������������就����øN��NGR�ĵõ�LPD-PEG-NGR��LPD-PEG-NGR��ȫ���ԡ������ԡ���Ч�ķ�ʽ��siRNA������С��ʵ�����ڣ�����ͨ������c-myc siRNA��ͨ���µ�c-myc�ı�����Ч��������ϸ���������Ӷ����Ʋ������������������⣬ͨ�������ɾ���ɸѡ�õ����������ģ�������Ч������ż����ͬ��ҩ��ҩ�������������֯�����Щ������������ĵ�ҩ�����ϵͳ�����������ƾ�����Ҫ��DZ����
�����ض��е��Ŀ�ԭ�Ŀ�����Ϊ���������磬ͨ���뿹ԭ�ʵ�ϸ���ϵ���Ҫ��֯�����Ը����� (MHC) ��ϣ�����������ϸ������ T ϸ���Ŀ��������á�EGFR������ EGFR1 �� HER2����������֪�İ�֢���ưе㡣���� HER2 �ṹ�������� TERT572Y ������ 46 �����ڷ�Сϸ���ΰ����ߡ�Ƥ��ע�� TERT572Y ���յ� TERT ���������߷�Ӧ�������ӳ������ڡ�Manijeh��ͨ��PEPOP�������㲢Ԥ��DZ�ڱ�λ����HER2��������ɸѡ��������������Ϊ��ѡ���У�ͨ�����ӶԽ�������Щ��ѡ����MHC I���II����ӵĽ��������Ѱ������MHC I���II��������ȶ��Ľ�Ͻṹ��ɸѡ��MHC I���II��������Ϊ���ٰ�������396��������������ٴ�����δ����ʾ�����������Ч����������Ҳ��˺����ܵ���ע��������Takumi������Ϊ���������ڵĴ������֢�������ٴ��о���δ�ܳɹ�����Ҫԭ����������ԭ�Խϲ�������Ż��ĵ��䷽��������ҩ;������������Ч�����ı���Ϊϸ�����ģ�CPP����Ҳ������Ϊҩ�����壬�������ġ������ʡ�DNA��СRNA��ҩ�����͵�ϸ���С���Nerinetide��CPP Tat��ɵ�CPP-ҩ�ﹹ���屻���ڽ�Nerinetide���ʹ���Ѫ�����ϲ�������Ԫ. �Ļ�ͨ���뿹ԭ������յ���ԭ�������������ܲ������Ѱз�Ӧ�ķ��գ���ʾ�����õĴ��ݹ��� . Tsoras ʹ�������״����������ǵ�λ������߿�ԭ������ԭ�� .
���ij�����Ϊҩ�����塢�����⣬������ͨ����������Ϸ��ӿ��������ã��������ܹ�ע�������PD-1/PD-L1�ź�ͨ·�Ķ��ġ�Boohaker�������һ��PD-L1��ģ����PL120131�����Ŀ���ͨ����PD-1���������PD-1/PD-L1������ã���3D������ģ���У�PL120131��PD-1�������ά�ֹ�����Tϸ���Ĵ��ͻ��ԡ�Zhou�Ȼ�����PD-1��PD-L1��ϵĶ��ģ��������������DS-I��DS-II���价����ʽ����PD-1���ֳ���ǿ��������Abbas ���������һ���µİ��� PD-1 �Ķ���FITC-YT-16���ö���������������ǿ��Tϸ���Ŀ��������ԣ��� Sasikumar ��������˶��� NP-12����PD-1�����Խ�� PD-L1�����⣬NP-12 �ں�ɫ�������᳦������ϸ�������ٴ�ǰģ���У�������ԭ����������������ת�Ʒ�����ֳ�����ҵ�� PD-1 ��������ͬ��Ч������Ȼ��Щ�Ļ����ʺ����PD-1/PD-L1�����������������Ǿ��й�����ǰ�������Զ���Ķ����ģ�VP��Ҳ���ܱ��ֳ����������á�����VP��Ȼ�����鶯�����壬���Ƕ�ϸ��Ĥ�ϵ��ض�����ͨ����������ֳ��߶ȵ������Ժ�ѡ���ԡ�������֩���з���������Ķ���Hanatoxin-1�������Ե������Ĥ�ϵ�K +ͨ�����ڽ᳦���ķ�չ�����й۲쵽��K +ͨ���ĸ߱��Okada���˷��ִ�Lachesana sp�д������¿���LaFr26. ֩�붾Һ�Էΰ�ϸ��ϵ LX22 �� BEN ��ϸ���������ã�������ϸ��������Դ�� K +������AMPs �������е�����Ҳ���������ǵĹ�ע��һЩ AMPs ��֤�����п��������ԣ�����һЩ��ٽ�������չ��Strzelecka ���˺ϳɵļ� ��-�������������� 3D ����ģ�������������ٰ�ϸ��������������� ��-��������������п���DZ�����������������������ӣ�ͨ���۵�������Ĥ��������ɱ����ϸ�����ο������ĵ�Ĥ����������ã�Aronson �����Ʊ���һ��������֬�ʿ��������ǿ���������ϸ��Ĥ�����ںϲ��鵼ϸ��ɱ�ˣ�������ϸ������û�ж��ԣ�������һ���µ������ܽ���ԡ�
��Ȼ����������ٴ�ǰ���ٴ��о��ж���ʾ�����õĿ�����Ч������Ŀǰֻ�����ֶ��ı������������������ֱ������ڹ���������Ī��º����ڶ�Թ������Ŀ������ף�����Էΰ���θ���ȸ����������������Զ��ĵ����Ʋ����о����ڽ����С���˹ؼ�����Ѱ�Ҹ���������ϸ���������Ա��������е㣬����ǿ��ҽѧת�������⣬��Ը�����������Ķ������Ҳ��һ��DZ�ڵIJ��ԡ�
��������
������������������������ࡢ���ֲ�ϸ����ϸ��������һֱ���ܲ����Լ���֮�࣬������������Ѫ�ȡ����кͻ��������ȱ���ۺ��������̲��������ܹ�ȥ��ʮ�����������ڿ�����ҩ���з����渶���˾�Ŭ�������ֿ�����ҩ���ѻ�����Ͷ���ٴ�ʹ�ã������̲���һЩ������Ȼû����Ч�����Ʒ�����
�������ĵ��о��ѳ�Ϊһ�����Ż��⣬��Ϊ����ҩ����и߶ȵ������Ժͻ��ԡ�����������Ҫͨ������������������ϸ�Ⱦ������Τ���ǵ�һ�������Ŀ������ģ���һ�� 36 ����������ģ���ͨ����� gp41��HIV ��Ĥ���ף��������ظ��ṹ������ֹ���ںϣ��Ӷ���� HIV ��Ⱦ��2011 �꣬��������ҩ�ﲩ����ά��������ά���������ٴ����Ʊ����ײ��� (HCV) �����Ǿ��� HCV NS3/4A ˿���ᵰ��ø��ϣ����Ƶ���ø���ԣ��Ӷ�����������ڵ� HCV ����. ������ڿ������ĺ�ѡҩ����о����ڽ����ٴ�ǰ���ٴ��о��У�������� HBV �� HDV �� myrcludex B ��������в����� flufirvitide ���Լ���� HIV-1 �� sifuvirtide��
2020������������״����SARS-CoV-2����ĺ������������Լ�������������ȫ�������ǵ������2020������鿪ʼ��������ѧ����Ͷ���˴����ľ����о�COVID-19�ĸ�Ⱦ���ƣ���Ѱ�ҿ�COVID-19�����Ʒ�����ҩ�����������ҩ�COVID - 19�����鱻���ٲ���Ϊһ���а�Ĥ������RNA��״�������������СԼΪ29.9kb���������״������SARS-CoV�����������
���类�ձ���Ϊ��Ԥ�����в���������Ч�ֶΣ�Ŀǰ����������һ���ʹ�ã���������mRNA������ �������ٲ����������硢�������ȡ�����������������Ըߡ���ȫ�Ժá������������ƣ���ΪSARS-CoV-2�����з���һ����Ծ���о������ڸ�Ⱦ���ƣ�����о�С����ƺ����������SARS-CoV-2�������硣��������������Ϣѧ����������ʶ����������ʶ��SARS-CoV-2��ͻ�ǵ���Bϸ����Tϸ���ؼ���λ��Li���˺�Chakraborty���˽���Ȼ��λ������Ϊ���SARS-CoV-2�������ѡ�� ����Bhattacharya���˺�Waqas���Ի�������Bϸ����Tϸ���ı�λƬ�ι����µ�����ΪCOVID-19�����ѡ�� �������о�С��Ҳ��չ�����ƵĹ�����Herst ���˳�������������������������� CTL �������о�ƽ̨��� COVID-19 �����磬�������һϵ���������ѡ����������о����������úϳ��Ļ��������� SARS-CoV-2 �ĸ�Ⱦ���̡�
�� COVID-19 �����ڼ䣬�������ĵĿ��������˹㷺��ע����������� SARS-CoV-2 ��������Ŀ�����������Ϣѧ���������ڱ�λ����ơ������ʶ��ͷ��ӶԽӵ��¼����ѱ�Ѹ��������ƺ�ʶ���������ѡ�������δ������������������� COVID-19������������Ŀ��������Ѿ������˱���ľ��飬��Щ���粻����� SARS-CoV-2���������δ�����²�����
������۵�
������������������ص��������Ժ�����DZ������Ϊһ����ص�����ҩ����ܶ�����ijЩ��������С���Ӻʹ���������Ƽ��������ڰ���������ھ����ԣ�������������Ĥ��ͨ�Ժ������ȶ��Բ�����⡣Ϊ�˿˷���Щȱ�㣬���ǶԶ���ҩ��ķ��֡��������Ż������˹㷺���о�����ͳ���ȵ��ķ��ַ����������ƺ��ɾ���չʾ���¼����Ľ�ϣ�Ϊ�ڶ�ʱ���ڿ�����Ч��ѡ���Ե��ȵ����ṩ�˿ɿ��ķ�������ѧ����������ϳɷ����ĵ���������ʹ�ÿ��Ը�Ч���ɿ��ش��ģ�����ϳ��ġ���Щ�Ŀ���ͨ����ѧ�ϳɻ��Ŵ�������չ��λ�������Է�ʽ��һ�����Σ�����ǿ���ȶ��Ժ��������ԡ�
��Ȼ������������ʼ����Ȼ���أ����˺��ֺͿ��������ѴӼ�ģ����Ȼ���ػ�������Ȼ�����ת��Ϊ������ƾ��������������������Ե��ġ���������ѧ���Ļ�ѧ���ĵ��ͼ������ش�ͻ��ʹ��ҩ��֡���������������Ӧ������ȡ�����ش��չ������Ϊֹ������ 80 �����������Ľ���ȫ���г��������������ڽ����ٴ�ǰ�о����ٴ���������Щ��ҩ���ѱ�Ӧ���ڹ㷺�ļ���������������Ѫ�ܼ�����θ������������֢����Ⱦ�����Լ����翪�������ǵ���������DZ�����г�ǰ���;��ü�ֵ������Ԥ���������Ľ���������Ͷ�ʺ��о����ȣ���ȡ�ó��ڳɹ���
��������������Ϊ��ҵ����ѧϰ����Ȩ��ԭ����ԭ��־���У�������Ȩ������ϵɾ�������±�ע���������³����������Ķ�ԭ�ļ��ο����ף����Ķ�ԭ��־��