еЊвЊЃКЕУвцгкаТЕФЩњВњЁЂаоЪЮКЭЗжЮіММЪѕЃЌыФРрвЉЮяЕФПЊЗЂдкЙ§ШЅЪЎФъжаШЁЕУСЫОоДѓНјеЙЁЃыФРрвЉЮяЕФЩњВњКЭаоЪЮВЩгУСЫЛЏбЇКЭЩњЮяЗНЗЈЃЌВЂВЩгУСЫаТгБЕФЩшМЦКЭЕнЫЭВпТдЃЌетгажњгкПЫЗўыФРрвЉЮяЕФЙЬгаШБЯнЃЌВЂЭЦЖЏИУСьгђЕФГжајЗЂеЙЁЃШЫУЧвбОЛёЕУВЂбаОПСЫЖржжЬьШЛКЭаоЪЮыФРрвЉЮяЃЌКИЧСЫЖрИіжЮСЦСьгђЁЃБОзлЪізмНсСЫыФРрвЉЮяЗЂЯжЁЂЩњВњКЭаоЪЮЗНУцЕФХЌСІКЭГЩОЭЃЌвдМАЫќУЧФПЧАЕФгІгУЁЃЮвУЧЛЙЬжТлСЫжЮСЦадыФРрвЉЮяЮДРДЗЂеЙЕФМлжЕКЭЬєеНЁЃ
жЮСЦадыФЪЧгЩвЛЯЕСагаађАБЛљЫсзщГЩЕФвЛРрЖРЬивЉЮяЃЌЗжзгСПЭЈГЃЮЊ500-5000DaЁЃжЮСЦадыФЕФбаОПЪМгкЖдвШЕКЫиЁЂДпВњЫиЁЂМгбЙЫиКЭДйадЯйМЄЫиЪЭЗХМЄЫи(GnRH)ЕШЬьШЛШЫЬхМЄЫиМАЦфдкШЫЬхжаЬиЖЈЩњРэЛюадЕФЛљДЁбаОП[2]ЁЃзд1921ФъКЯГЩЕквЛИіжЮСЦадыФвШЕКЫивдРДЃЌвбОШЁЕУСЫСюШЫжѕФПЕФГЩОЭЃЌШЋЧђвбга80ЖржжыФРрвЉЮяЛёХњЁЃвђДЫЃЌыФРрвЉЮяЕФПЊЗЂвбГЩЮЊвЉЮябаОПЕФзюШШУХЛАЬтжЎвЛЁЃ
20ЪРМЭЩЯАывЖЃЌШЫУЧЗЂЯжСЫМИжжПЩвдеќОШЩњУќЕФЩњЮяЛюадыФЃЌР§ШчвШЕКЫиКЭДйЩіЩЯЯйЦЄжЪМЄЫиЃЌетаЉыФзюГѕЖМЪЧДгЬьШЛРДдДжабаОПКЭЗжРыГіРДЕФЁЃвШЕКЫиЪЧвЛжжКЌга51ИіАБЛљЫсЕФыФЃЌЫќЕФЗЂЯжКЭПЊЗЂБЛШЯЮЊЪЧвЉЮяЗЂЯжСьгђЕФжиДѓПЦбЇГЩОЭжЎвЛЁЃЫќгк1921ФъгЩFrederickBantingЪзДЮЗжРыГіРДЃЌВЂгЩFrederickКЭCharlesBestНјвЛВНПЊЗЂ[3,4]ЃЌдкЪзДЮЗжРыКѓНівЛФъБуПЩгУгкжЮСЦЬЧФђВЁЛМепЁЃ1923ФъЃЌвШЕКЫиГЩЮЊЕквЛжжЩЬвЕЛЏыФРрвЉЮяЃЌЦљНёвбдьИЃЪ§ЧЇУћЬЧФђВЁЛМепЁЃШЛЖјЃЌ20ЪРМЭШЫРрвШЕКЫиЕФВњСПЮоЗЈТњзуЪаГЁашЧѓЃЌХЃвШЕКЫиКЭжэвШЕКЫиЕШЖЏЮядДадвШЕКЫидквШЕКЫиЪаГЁЩЯеМОнжїЕМЕиЮЛНќ90ФъЃЌжБЕНБЛжизщвШЕКЫиШЁДњЁЃ
20ЪРМЭ50ФъДњжС90ФъДњЃЌИќЖрОпгажЮСЦЧБСІЕФыФМЄЫиМАЦфЪмЬхЯрМЬБЛМјЖЈКЭБэеї7ЁЃЭЌЪБЃЌЕААзжЪДПЛЏКЯГЩЁЂНсЙЙНтЮіКЭВтађММЪѕШЁЕУЪЕжЪадНјеЙЃЌМгЫйСЫыФРрвЉЮяЕФбаЗЂЃЌШЋЧђвбгаНќ40жжыФРрвЉЮяЛёХњЁЃжЕЕУзЂвтЕФЪЧЃЌГ§СЫЬьШЛыФЭтЃЌКЯГЩДпВњЫиЁЂКЯГЩМгбЙЫиЁЂжизщШЫвШЕКЫиЕШКЯГЩыФвВПЊЪМБЛПЊЗЂЁЃ
НјШы21ЪРМЭЃЌЖрыФвЉЮябаЗЂНјШыаТМЭдЊЃЌНсЙЙЩњЮябЇЁЂжизщЩњЮяжЦМСЁЂаТаЭКЯГЩЗжЮіММЪѕЕФНјВНДѓДѓМгЫйСЫЖрыФвЉЮябаЗЂНјГЬЃЌвбНЈСЂСЫАќРЈЖрыФвЉЮяЗЂЯжЁЂвЉЮяЩшМЦЁЂЖрыФКЯГЩЁЂНсЙЙаоЪЮЁЂЛюадЦРМлЕШЛЗНкЕФЖрыФвЉЮябаЗЂЬхЯЕЁЃзд2000ФъвдРДЃЌШЋЧђЙВХњзМ33жжЗЧвШЕКЫиЖрыФвЉЮяЃЈБэ1ЃЉЁЃДЫЭтЃЌетаЉЖрыФвЉЮявбВЛдйЪЧМђЕЅЕФМЄЫиФЃФтЮяЛђгЩЬьШЛАБЛљЫсзщГЩЁЃР§ШчЃЌЖїЗђЮЄыФЪЧвЛжж36ИіАБЛљЫсЕФЗТЩњыФЃЌФЃФтШЫРрУтвпШБЯнВЁЖОЃЈHIVЃЉЕААзЃЌгУгкСЊКЯжЮСЦHIV-1 ;ЦыПМХЕыФЪЧвЛжжДггѓТнConusmagusжаЬсШЁЕФЩёОЖОадыФЃЌгк2004ФъЛёХњЃЌгУгкжЮСЦбЯжиТ§адЬлЭДЃЛЬцЖШТГыФЪЧвЛжжвШИпбЊЬЧЫибљыФ2(GLP-2)РрЫЦЮяЃЌгУгкжЮСЦЖЬГІзлКЯеїЃЌВЩгУжизщDNAММЪѕИФдьЕФДѓГІИЫОњОњжъжЦдьЃЛРћРТГыФЪЧШЫРрвШИпбЊЬЧЫибљыФ1(GLP-1)ЕФЛЏбЇКЯГЩРрЫЦЮяЃЌЭЈЙ§НЋC-16жЌЗОЫсЃЈзищЕЫсЃЉгыРЕАБЫсВаЛљЃЈађСажаЕФЮЛжУ26ЃЉЩЯЕФЙШАБЫсМфИєЮяСЌНгЖјГЩЃЌПЩзїЮЊGLP-1ЪмЬхМЄЖЏМСРДжЮСЦ2аЭЬЧФђВЁ(T2DM)ЁЃЫљгаетаЉыФРрвЉЮяЖМвбгУгкЙуЗКЕФжЮСЦСьгђЃЌР§ШчУкФђПЦЁЂКєЮќПЦЁЂЬлЭДПЦЁЂжзСіПЦЁЂДњаЛПЦЁЂаФбЊЙмПЦКЭПЙОњПЦЁЃЦљНёЮЊжЙЃЌвбга170ЖржжыФРрвЉЮяДІгкЛюдОЕФСйДВПЊЗЂНзЖЮЃЈБэ2ЃЉЃЌЛЙгаИќЖрыФРрвЉЮяДІгкСйДВЧАбаОПНзЖЮЁЃ
ЖрыФвЉЮядквНвЉЪаГЁжаеМгаЯрЕБДѓЕФБШжиЃЌ2019ФъШЋЧђЯњЪлЖюГЌЙ§700вкУРдЊЃЌНЯ2013ФъдіГЄСНБЖЖрЁЃОнNjardarsonЕШЭГМЦЃЌ2019ФъЯњЪлЖюХХУћЧА200ЕФвЉЮяжаЃЌга10жжЗЧвШЕКЫиЖрыФвЉЮяЁЃгавтЫМЕФЪЧЃЌЖрыФвЉЮяЯњЪлЖюЧАШ§ЕФОљЮЊжЮСЦ2аЭЬЧФђВЁЕФGLP-1РрЫЦЮяЃЌАќРЈХХУћЕк19ЮЛЕФTrulicityЃЈЖШРЬЧыФЃЉЃЌСуЪлЖю43.9вкУРдЊЃЌХХУћЕк32ЮЛЕФVictozaЃЈРћРТГыФЃЉЃЌЯњЪлЖю32.9вкУРдЊЃЌХХУћЕк83ЮЛЕФRybelsusЃЈЫїТэТГыФЃЉЃЌЯњЪлЖю16.8вкУРдЊЃЈЭМ1ЃЉЁЃ
БОЮФЛиЙЫСЫыФРрвЉЮяЕФРњЪЗЗЂеЙКЭыФРрвЉЮяЗЂЯжЕФзюаТНјеЙЁЃЮвУЧжиЕуЙизЂжЮСЦадыФЕФвЉРэЬиадЃЌВЂжиЕуНщЩмИФНјыФРрвЉЮяЩшМЦЁЂКЯГЩЁЂаоЪЮКЭЦРЙРЕФаТММЪѕЃЌВЂЮЊыФРрвЉЮяЕФгІгУЬсЙЉаТЕФЪгНЧЁЃЮвУЧЛЙНЈвщЖСепВЮдФзюНќЕФМИЦЊзлЪівдЙЉНјвЛВНдФЖС.
жЮСЦадыФЃКгХЕуКЭШБЕу
жЮСЦадыФЭЈГЃгУзїМЄЫиЁЂЩњГЄвђзгЁЂЩёОЕнжЪЁЂРызгЭЈЕРХфЬхЛђПЙИаШОМСЁЃЫќУЧгыЯИАћБэУцЪмЬхНсКЯВЂвдИпЧзКЭСІКЭЬивьадв§ЗЂЯИАћФкаЇгІЃЌзїгУЗНЪНгыЩњЮяжЦМСЃЈАќРЈжЮСЦадЕААзжЪКЭПЙЬхЃЉЯрЫЦЁЃШЛЖјЃЌгыЩњЮяжЦМСЯрБШЃЌжЮСЦадыФЕФУтвпдадНЯЕЭЃЌЩњВњГЩБОНЯЕЭ.
аЁЗжзгвЉЮяОпгагЦОУЕФжЮСЦРњЪЗЃЌОпгаЩњВњГЩБОКЭЯњЪлМлИёЕЭЁЂПкЗўЁЂФЄДЉЭИадКУЕШЙЬгагХЪЦЁЃгыыФКЭЩњЮяжЦМСЃЈЕААзжЪЛђПЙЬхЃЉЯрБШЃЌЬьШЛЬсШЁКЭЛЏбЇКЯГЩЕФаЁЗжзгОљОпгаОКељадЕФМлИёгХЪЦЁЃПкЗўаЁЗжзгОпгаИќКУЕФАВШЋадКЭЬсИпЛМепвРДгадЕФКУДІЃЌЭЌЪБЫќУЧЕФаЁГпДчвВЪЙЫќУЧФмЙЛДЉЭИЯИАћФЄвдАаЯђЯИАћФкЕФЗжзгЁЃШЛЖјЃЌЫќУЧЕФаЁГпДчвВвтЮЖзХЫќУЧКмФбгааЇЕивжжЦДѓБэУцЯрЛЅзїгУЃЌР§ШчЕААзжЪ-ЕААзжЪЯрЛЅзїгУЃЈPPIЃЉЁЃPPI ЭЈГЃеМОн 1500ЈC3000 A2ЕФНгДЅУцЛ§ЃЌЖјаЁЗжзггЩгкЦфЗжзгГпДчгаЯоЃЌжЛФмИВИЧ 300ЈC1000 A2ЕФЕААзжЪБэУцЁЃЯрБШжЎЯТЃЌыФРрвЉЮяЖРЬиЕФЮяРэЛЏбЇаджЪЃЌАќРЈЦфИќДѓЕФГпДчКЭИќСщЛюЕФЙЧМмЃЌЪЙЫќУЧФмЙЛзїЮЊ PPI ЕФЧПаЇвжжЦМС,аЁЗжзгвЉЮяЕФСйДВгІгУвВвђЬивьадЕЭгкЖрыФРрвЉЮяЖјЪмЕНЯожЦЃЌР§ШчЫїРЗЧФсКЭЪцФсЬцФсЖМЪЧРвАБЫсМЄУИвжжЦМСЃЌПЩвжжЦбЊЙмФкЦЄЩњГЄвђзгЃЈVEGFЃЉЪмЬхЕФРвАБЫсМЄУИНсЙЙгђЛюадЃЌВњЩњПЙбЊЙмЩњГЩзїгУЃЌгУгкжЮСЦАЉжЂЛМепЃЛШЛЖјЃЌЫќУЧвВАаЯђЦфЫћМЄУИЪмЬхЃЌШчЫПАБЫс/ЫеАБЫсМЄУИЪмЬхЃЌЕМжТЯИАћЖОад.
зїЮЊЛљгкЬьШЛАБЛљЫсЕФжЮСЦМСЃЌжЮСЦадыФОпгаСНИіЙЬгаШБЕуЃЈЭМ2ЃЉ ЃКФЄВЛЭЈЭИадКЭЬхФкЮШЖЈадВюЃЌетЪЧыФвЉЮяПЊЗЂЕФжївЊАэНХЪЏ.
1ЁЂЖрыФЕФФЄЭЈЭИадНЯШѕЃЌЖрыФвЉЮяЕФФЄЭЈЭИадШЁОігкЖржжвђЫиЃЌАќРЈЖрыФГЄЖШЁЂАБЛљЫсзщГЩЕШЁЃЖрыФвЛАуЮоЗЈДЉЙ§ЯИАћФЄЕНДяЯИАћФкАаЕуЃЌвђДЫЯожЦСЫЦфдквЉЮяПЊЗЂжаЕФгІгУЁЃ2018ФъLauЕШБЈЕРЃЌСйДВПЊЗЂжа90%вдЩЯЕФЖрыФАаЯђЯИАћЭтАаЕуЃЌАќРЈGЕААзХМСЊЪмЬхЃЈGPCRsЃЉЁЂДйадЯйМЄЫиЪЭЗХМЄЫиЃЈGnRHЃЉЪмЬхЁЂвШИпбЊЬЧЫибљыФ1ЃЈGLP-1ЃЉЪмЬхЁЃ
2ЁЂыФдкЬхФкЕФЮШЖЈадНЯВюЁЃЬьШЛыФгЩЭЈЙ§ѕЃАЗМќСЌНгЕФАБЛљЫсСДзщГЩЃЌЕЋШБЗІЖўМЖЛђШ§МЖНсЙЙЫљИГгшЕФЮШЖЈадЁЃдкУЛгаШЮКЮБЃЛЄЕФЧщПіЯТЃЌЕББЉТЖгкЛЗОГжаЪБЃЌѕЃАЗМќКмШнвзБЛЬхФкУИЫЎНтЛђЦЦЛЕЁЃетаЉЙЬгаЕФЛЏбЇаджЪЪЙыФдкЛЏбЇКЭЮяРэЩЯВЛЮШЖЈЃЌАыЫЅЦкЖЬЃЌЬхФкЯћГ§ПьЁЃ
ЖрыФЕФетаЉФкдкгХЪЦгыСгЪЦМШЮЊЖрыФвЉЮяПЊЗЂДјРДСЫЬєеНЃЌвВЮЊЖрыФвЉЮяЕФЩшМЦКЭгХЛЏДјРДСЫЛњгіКЭЗНЯђЁЃ
жЮСЦадыФЕФЗЂеЙТЗОЖЃКЗЂЯжЁЂЩњВњКЭгХЛЏ
ыФРрвЉЮябаЗЂ
ШЫЬхФкЕФЬьШЛыФ/МЄЫи
ыФРрвЉЮябаЗЂЕФРњЪЗЪМгкРћгУОпгаГфЗжбаОПЕФЩњРэЙІФмЕФЬьШЛМЄЫиКЭыФРрРДжЮСЦвђМЄЫиШБЗІв§Ц№ЕФМВВЁЃЌР§Шч 1 аЭЬЧФђВЁЛђ 2 аЭЬЧФђВЁЛМепШБЗІЕїНкбЊЬЧЫЎЦНЫљашЕФвШЕКЫиЁЃЬЧФђВЁЕФжЮСЦЗНЗЈЪЧзЂЩфвШЕКЫиЃЌЛђДЬМЄвШЕКЫиЗжУкЯрЙиАаЕуЃЈШч GLP-1 ЪмЬхЃЉВњЩњвШЕКЫиЁЃбАевЬьШЛыФРрКЭМЄЫиЛђгУЖЏЮяЭЌдДЮяЃЈШчвШЕКЫиЁЂGLP-1ЁЂЩњГЄвжЫиЁЂGnRHЁЂ8-Arg-МгбЙЫиКЭДпВњЫиЃЉЬцДњЫќУЧЃЌетЪЧыФРрвЉЮябаЗЂЕФзюГѕВпТдЃЈЭМ3ЃЉ)ЁЃШЛЖјЃЌетаЉЬьШЛыФЫљДцдкЕФШБЕув§Ц№СЫШЫУЧЖдгХЛЏЦфЬьШЛађСаЕФаЫШЄЃЌДгЖјВњЩњСЫвЛЯЕСаЬьШЛМЄЫиФЃФтыФвЉЮяЁЃ
МЄЫиФЃФтыФ
GLP-1бмЩњЕФЖрыФвЉЮяЃЈЭМ4aЃЉЃКGLP-1ЪЧвЛжж37ИіАБЛљЫсЕФЖрыФЃЌПЩЕїНквШЕКЫиЕФВњЩњКЭЗжУкЃЌдкЬхФкЕФАыЫЅЦкКмЖЬЁЃШЫУЧИЖГіСЫДѓСПЕФХЌСІРДаоИФЦфађСаЃЌвддіЧПетжжМЄЫиЕФЮШЖЈадЃЌЭЌЪББЃГжЦфаЇСІКЭвЉРэзїгУЃЌДгЖјПЊЗЂГіШ§жжзюГЉЯњЕФПЙ2аЭЬЧФђВЁЖрыФвЉЮяЃКTrulicityЃЈЖШРЬЧыФЃЉ ЁЂVictozaЃЈРћРТГыФЃЉКЭOzempicЃЈЫїТэТГыФЃЉЁЃ
ДйадЯйМЄЫиЪЭЗХМЄЫи (GnRH) бмЩњЕФыФвЉЮяЃЈЭМ4bЃЉЃКGnRH ЪЧвЛжжКЌга 10 ИіАБЛљЫсЕФыФЃЌгЩЯТЧ№ФджаЕФ GnRH ЩёОдЊВњЩњЁЃЖд GnRH ЬьШЛађСаЕФаоИФвбЕМжТМИжжыФвЉЮяЕФПЊЗЂЃЌР§ШчССБћШ№СжКЭЕиМгШ№ПЫЁЃССБћШ№СжЭЈЙ§МЄЛю GnRH ЪмЬхОпгагы GnRH ЯрЭЌЕФЩњЮяЛюадЃЌВЂгУзї GnRH ЪмЬхМЄЖЏМСЃЌгУгкжЮСЦМЄЫиЗДгІадЧАСаЯйАЉЁЂзгЙЌФкФЄвьЮЛжЂЁЂзгЙЌМЁСіКЭаддчЪьЁЃЫфШЛЕиМгШ№ПЫЕФађСаЪЧДг GnRH гХЛЏЖјРДЕФЃЌЕЋЫќЭЈЙ§ОКељадЕигы GnRH ЪмЬхНсКЯГфЕБ GnRHозПЙМСЃЌгУгкжЮСЦЭэЦкЧАСаЯйАЉЁЃ
аэЖрЦфЫћвбХњзМЕФыФРрвЉЮявВРДдДгкЬьШЛМЄЫи1ЃЌАќРЈАТЧњыФЃЌвЛжжЩњГЄвжЫиФЃФтыФРрвЉЮяЃЌгУгкжЮСЦВњЩњЩњГЄМЄЫиЕФжзСіКЭДЙЬхСіЃЛШЅАБМгбЙЫиЃЌвЛжж 8-Arg-МгбЙЫиФЃФтыФРрвЉЮяЃЌгУгкжЮСЦФђБРжЂКЭвЙФђжЂЃЛПЈБДЫѕЙЌЫиЃЌвЛжжгУгкжЮСЦБеОЕФДпВњЫиЭЌдДЮяКЭАЂЭаЮїАрЃЌвЛжжгУгквжжЦдчВњЕФДпВњЫиозПЙМСЁЃ
ДгЬьШЛВњЮяжаМјЖЈЕФыФ
РДздЯИОњЁЂецОњЁЂжВЮяКЭЖЏЮяЕФаэЖрЩњЮяЛюадыФЖМОпгажЮСЦЬиадЃЌР§ШчЩпЖОЃЌЫќБЛШЯЮЊЪЧбЊЙмФкЦЄЩњГЄвђзг(VEGF)РрЫЦЮяЃЌVEGF-FЛђsvVEGFЁЃЫќУЧЭЈГЃЪЧЖўСђМќЗсИЛЕФЛЗыФЃЌВаЛљВЛГЌЙ§80ИіЃЌПЩЭЈЙ§АаЯђРызгЭЈЕРКЭЦфЫћФЄНсКЯЪмЬхгеЕМЯИАћЖОадЁЃРДздЩпКЭаЋзгЕФЖОвКыФвбБЛаоЪЮгУгкжЮСЦгІгУЁЃДЫЭтЃЌДгGilaЙжЮяЖОвКжагХЛЏЕФАЌШћФЧыФЃЈЭМ5aЃЉЪЧвЛжжGLP-1МЄЖЏМСЃЌЖјЦыПМХЕыФЪЧвЛжжРДздгѓТнЕФЖОвКыФЃЌвбБЛгУгкжЮСЦТ§адЩёОадЬлЭД
ЗЧКЫЬЧЬхыФЃЈNRPЃЉЪЧДгЬьШЛВњЮяжаМјЖЈЕФСэвЛРрыФЁЃађСажаКЌгаЕФЗЧБъзМВаЛљвтЮЖзХNRPВЛЪЧЭЈЙ§ДЋЭГЕФКЫЬЧЬхЩњЮяКЯГЩЭООЖВњЩњЕФЃЌЖјЪЧгЩЗЧКЫЬЧЬхыФКЯГЩУИЭЈЙ§гЩЦ№ЪМЁЂбгГЄКЭжежЙФЃПщзщГЩЕФЭООЖВњЩњЕФЁЃгыКЫЬЧЬхКЯГЩЕФыФЯрБШЃЌNRPЖдЫЎНтУИЕФЕжПЙСІИќЧПЃЌдкЬхФкЕФЮШЖЈадвВИќИпЁЃбаОПзюЖрЕФNRPжївЊРДдДгкЯИОњКЭецОњЃЌАќРЈЭђЙХУЙЫиЁЂЛЗцпОњЫиЁЂТЗЕЫУЙЫиЁЃЃЈЭМ5b)ЁЂОпгаПЙОњЛюадЕФЬцПЫЫїАЭЖЁЃЌвдМАОпгаПЙжзСіЛюадЕФa-ЖьИроІМюЁЂФЩУзФвЫиAКЭЗХЯпОњЫи73ЁЂ74 ЁЃДЫЭтЃЌЛЗыФЪЧЛЗзДыФЃЌАќКЌЭЈГЃдкжВЮяжаМјЖЈЕФвЛРрЬиЖЈNRP 75ЁЂ76ЁЂ77 ЃЌР§ШчЖїСОњЫиBКЭАЌФЊЕТЦеЫЙЕТ78ЁЂ79ЁЃетаЉыФвЉЮяБэЯжГідіЧПЕФбЊНЌЮШЖЈадЃЌЪЙЦфФмЙЛПкЗўИјвЉЁЃШЛЖјЃЌNRPЕФКЯГЩКЭЙЙаЇЙиЯЕбаОПЪЧNRPзюОпЬєеНадКЭзюСюШЫаЫЗмЕФбаОПСьгђжЎвЛЁЃ
ИљОнЕААзжЪ-ЕААзжЪЯрЛЅзїгУКЯРэЩшМЦыФ
ЕААзжЪзщбЇКЭНсЙЙЩњЮябЇЕФЗЂеЙЕМжТЗЂЯжСЫаэЖрВЮгыДѓЖрЪ§ЯИАћЙ§ГЬКЭЩњЮяЙІФмЕФЕААзжЪ-ЕААзжЪЯрЛЅзїгУ(PPI) ЁЃЦљНёЮЊжЙЃЌвббаОПСЫГЌЙ§14,000жжPPIЃЌНіеМШЫЬхЫљгаPPIЕФ1%зѓгвЁЃPPIЛЙЕїНкШЫРрМВВЁжаЕФаэЖрживЊЯИАћЭЈТЗЃЌвђДЫЪЧЧБдкЕФвЉЮяАаЕуЖрыФзїЮЊPPIвжжЦМСЛђМЄЖЏМСгыаЁЗжзгЁЂПЙЬхЕШЯрБШОпгаЯШЬьгХЪЦЃЌвђДЫЛљгквбжЊPPIОЇЬхНсЙЙЗЂеЙСЫвЛжжаТЕФЖрыФвЉЮяЗЂЯжММЪѕЃКЖрыФЕФКЯРэЩшМЦЃЌБЛШЯЮЊЪЧвЛжжКмгаЧАЭОЕФЗЂЯжаТаЭЖрыФвЉЮяКђбЁЮяЕФВпТдЁЃ
ыФЕФКЯРэЩшМЦЩцМАЛљгкФПБъPPIНтЮіОЇЬхНсЙЙЕФМЦЫуЛњИЈжњЩњЮяаХЯЂбЇММЪѕЁЃPPIНсКЯНчУцЕФЩњЮяаХЯЂбЇКЭМЦЫуЗжЮіПЩвдЪЖБ№СНжжЯрЛЅзїгУЕААзжЪБэУцЕФБиашАБЛљЫсЁЃетаЉБиашАБЛљЫсЙБЯзСЫPPIЕФжївЊМЊВМЫЙФмЃЌЭЈГЃГЦЮЊЁАШШЕуЁБ ЁЃШШЕуПЩФмЪЧЕААзжЪЕФСЌајЦЌЖЮЃЌвВПЩФмЪЧЕААзжЪВЛЭЌЖўМЖНсЙЙЩЯЕФЗжЩЂВаЛљЁЃPPIыФЕїНкМСЕФЩшМЦЛљгкетаЉШШЕуЃЌвЊУДжБНгЪЙгУСЌајЦЌЖЮЃЌвЊУДЪЙгУНЋЗжЩЂВаЛљСЌНгЦ№РДзїЮЊГѕЪМађСаЕФВпТдЁЃШЛЖјЃЌЮЊСЫЬсИпЦфЛюадКЭЮяРэЛЏбЇаджЪЃЌашвЊНјвЛВННјааыФЕФПЊЗЂКЭНсЙЙгХЛЏЃЌАќРЈыФЛЗЛЏКЭжїСДаоЪЮЁЃР§ШчЃЌЭЈЙ§баОПНсЙЙ-ЛюадЙиЯЕШЗЖЈБиашыФВаЛљВЂЬсГіЗЧБиашВаЛљЕФЬцДњЃЌвдМАЖдађСаНјааЛЏбЇаоЪЮвдЮШЖЈыФЖўМЖНсЙЙЃЌАќРЈзЊНЧЁЂТна§ЁЂЗЂМаКЭбгЩьЙЙЯѓЃЌПЩвддіЧПЩњЮяЛюадВЂИФЩЦЮяРэЛЏбЇаджЪЁЃ
ЭЈЙ§ЪЩОњЬхеЙЪОЗЂЯжыФРрКђбЁвЉЮя
ЪЩОњЬхеЙЪОЪЧвЛжжИпаЇЁЂПЩППЕФММЪѕЃЌгУгкЪЖБ№ЩњЮяАаБъЕФХфЬхЃЌгЩ Smith гк 1985 ФъЪзДЮБЈЕРЁЃЪЩОњЬхеЙЪОРћгУжизщММЪѕдкЪЩОњЬхБэУцЩшМЦАаБъХфЬхЁЃЪЩОњЬхжажЛВњЩњКЌгаЕААзжЪАБЛљЫсЕФыФЃЌЖјВЛЪЧ NRPЁЃетжжИпЭЈСПВтађЗНЗЈПЩгУгкЪЖБ№вЉЮяЯпЫїЃЌАќРЈПЙЬхКЭыФЁЃЪЩОњЬхеЙЪОвбЙуЗКгУгкЗЂЯжаТЕФыФХфЬхЁЃLerner ЕШШЫБЈЕРЭЈЙ§ЪЩОњЬхеЙЪОЗЂЯжСЫ GLP-1 КЭЦфЫћФЄЪмЬхХфЬхЕФЧПаЇыФРрЫЦЮяЃЌАќРЈЕААзжЪЁЂыФКЭЖОвКЃЌжївЊзїЮЊМЄЖЏМСЁЃДЫЭтЃЌАаЯђзЊЛЏЩњГЄвђзг (TGF)-ІТ1 ЛђБэЦЄЩњГЄвђзгЪмЬх (EGFR) ЕФыФЃЌвдМАЦЦЛЕГЩЯЫЮЌЯИАћЩњГЄвђзг (FGF)-1-FGFR1 ЯрЛЅзїгУЕФыФозПЙМСЪЧЭЈЙ§ЪЩОњЬхеЙЪОЗЂЯжЕФыФвЉЮяЕФСМКУР§згЁЃЪЩОњЬхеЙЪОММЪѕЕФзюаТЗЂеЙВржигкбАевИќгааЇЕФЩИбЁЗНАИЃЌвдМђЛЏКЃСПЪ§ОнжаЕФХфЬхбЁдёЃЌР§ШчЭЈЙ§МѕЩйЪЩОњЬхЬдбЁжмЦкЁЃHeinis ЕШШЫРћгУЁАЪЩОњЬхЩЯЁБаоЪЮММЪѕДгДЋЭГЪЩОњЬхеЙЪОжаЛёЕУЛЏбЇаоЪЮыФЃЌЛёЕУЫЋСђУбЛЗыФЁЃСэвЛжжВпТдЩцМАПЊЗЂаТгБЕФеЙЪОЗНЗЈЁЃР§ШчЃЌSchumacher ЕШШЫПЊЗЂСЫОЕЯёЪЩОњЬхеЙЪОРДЬНЫї D ЪжадыФЃЌЖј Szostak ЕШШЫНјаа mRNA еЙЪОвдЗЂЯжКЭбЁдёОпгаЗЧЬьШЛАБЛљЫсЕФДѓЛЗыФЁЃSuga ЕШШЫРћгУКЫЬЧЬхеЙЪОРДПЊЗЂКЌга D АБЛљЫсКЭЗЧЬьШЛАБЛљЫсЕФЯШЕМыФЃЌАќРЈЩњЮяЛюадДѓЛЗыФ.етаЉЗЂеЙЪЙЕУШЫУЧФмЙЛЙЙНЈДѓСПЕФеЙЪОЮФПтЃЌвдЗЂЯжаТЕФКђбЁыФЁЃ
жЮСЦадыФЕФКЯГЩгыаоЪЮ
ЗЂЯжОпгажЮСЦЧБСІЕФыФЪЧыФвЉЮяПЊЗЂЕФЕквЛВНЃЌжЎКѓЭЈЙ§ЛЏбЇЛђЩњЮяКЯГЩыФВЂНјааађСааоЪЮвдИФЩЦЦфвЉРэЬиадЁЃетРяЮвУЧзмНсСЫыФЩњВњКЭаоЪЮЫљгУЕНЕФЛљБОММЪѕЁЃ
ыФЕФЛЏбЇКЯГЩ
ыФЕФЛЏбЇКЯГЩММЪѕвбОКмЗЂДяЃЌЬиБ№ЪЧ1963ФъMerrifieldПЊЗЂЕФЙЬЯрыФКЯГЩЃЈSPPSЃЉММЪѕ.SPPSММЪѕдкЗНЗЈТлКЭКЯГЩВФСЯЗНУцЕУЕНСЫЯджјИФНјЃЌВЂдкЯжДњЖрыФЩњВњжаЗЂЛгзХжСЙиживЊЕФзїгУЁЃЫќЭЈЙ§НЋАБЛљЫсХМСЊКЭЭбБЃЛЄНсКЯдквЛИіМђЕЅЕФЗДгІЦїжаРДДйНјЖрыФЕФКЯГЩЃЌетНјвЛВНЕМжТСЫздЖЏЖрыФКЯГЩвЧЕФЗЂУїЁЃгыжизщММЪѕЯрБШЃЌSPPSЛёЕУЕФДжыФИќМгЕЅЕїЃЌВЛКЌЦфЫћЩњЮяЛЏКЯЮяЃЌШчУИЁЂDNAКЭRNAЦЌЖЮЁЂВЛЯрЙиЕФЕААзжЪКЭЖрыФЁЃДЫЭтЃЌзюжеSPPSВњЦЗжаЕФдгжЪКмШнвзЪЖБ№ЃЌвђЮЊЫќУЧжївЊРДздКЯГЩЙ§ГЬжаЕФВЛЭъШЋЗДгІЛђИБЗДгІЃЌЪЙЕУКѓајЕФДПЛЏЯрЖдМђЕЅЁЃ
SPPSАќРЈНЋАБЛљЫсЕФєШЛљХМСЊЕНЙЬЬхОлКЯЮяЪїжЌЩЯЃЌВЂНЋАЗЛљДгБЃЛЄЛљжаЪЭЗХГіРДЕФвЛИібЛЗЃЈЭМ6ЃЉЁЃИїжжЪїжЌЃЌШч4-МзЛљЖўБНМзЛљАЗ(HMBA)ЪїжЌЁЂWangЪїжЌЁЂ2-ТШШ§БНМзЛљТШ(CTC)ЪїжЌКЭMerrifieldЪїжЌЃЌгУгкНЋѕЃАЗЛђгЮРыєШЛљв§ШыыФЕФCЖЫЁЃЯжДњыФЙЄвЕЭЈЙ§НЋЪїжЌгыВЛЭЌЕФНгЭЗХМСЊЃЌПЊЗЂСЫИїжжЙІФмадЪїжЌЃЌДгЖјФмЙЛдкЙЬЯржаКЯГЩГЄыФКЭНјааыФЛЗЛЏЁЃдкКЯГЩЙ§ГЬжаЃЌАБЛљЫсЕФАЗЛљКЭВрСДЭЈГЃЪмЕНВЛЭЌЛЏбЇЛљЭХЕФБЃЛЄЃЌетЛсЕМжТыФОлМЏВЂНЕЕЭДжыФЕФДПЖШЁЃФПЧАвбПЊЗЂГіСНжжжївЊЕФSPPSВпТдЃКFmoc-SPPSКЭBoc-SPPSЃЌЗжБ№гУгкШЅГ§жївЊЕФАЗБЃЛЄЛљЭХЃЌМДмЬМзбѕєЪЛљЃЈFmocЃЉКЭЪхЖЁбѕєЪЛљЃЈBoc ЃЉЁЃ
Boc-SPPSЪЙгУШ§ЗњввЫсШмвКШЅГ§АЗБЃЛЄЛљЃЌЪЙгУЧтЗњЫсШмвКСбНтзюжеЕФыФЃЌЕЋетаЉЙ§ГЬАщЫцзХДЬМЄадЦјЮЖКЭЖОадЁЃFmocПЩвддкИќЮТКЭЕФЬѕМўЯТШЅГ§ЃЌвђДЫЭЈГЃЪзбЁFmoc-SPPSВпТдЁЃШЛЖјЃЌBoc-SPPSЖдгкГЄыФКЯГЩгагХЪЦЃЌвђЮЊШ§ЗњввЫсЭбБЃЛЄПЩвдгааЇЦЦЛЕыФКЯГЩЙ§ГЬжаЕФОлМЏЁЃFmoc-SPPSбаОПФПЧАжївЊМЏжадкНтОіСНИіжївЊЮЪЬтЃЌАќРЈГЄыФКЯГЩЙ§ГЬжаЕФОлМЏКЭФГаЉађСаЕФЬьЖЌѕЃбЧАЗЕФаЮГЩЁЃвбВЩгУЖржжЗНЗЈЃЌАќРЈгІгУЕЭШЁДњЖШЪїжЌЗжРыыФСДЁЂЮЂВЈМѕЩйЗДгІЪБМфЁЂЪЙгУЛьКЯШмМСзїЮЊЗДгІШмвКЁЂвдМАЪЙгУМйИЌАБЫсДђЖЯжїСДЕФHМќвдБмУтЛђМѕЩйSPPSЙ§ГЬжаЕФОлМЏЁЃFmoc-SPPSЙ§ГЬжаЬьЖЌѕЃбЧАЗЕФаЮГЩЯджјНЕЕЭСЫДжыФЕФДПЖШЁЃМѕЩйЬьЖЌѕЃбЧАЗаЮГЩЕФНтОіЗНАИЪЧЪЙгУЮЂВЈМѕЩйЗДгІЪБМфЃЌЛђЪЙгУN-ІС-ЭщЛљAspЈCGlyЖўыФ ЃЌЛђдкЭбБЃЛЄЙ§ГЬжаЬэМг1-єЧЛљБНВЂШ§пђЃЈHOBtЃЉЁЂOxymaPureЁЃ
Fmoc-SPPSКЯГЩЩйгк50ИіВаЛљЕФыФЯрЖдГЃЙцЃЌЕЋНЯГЄыФЃЈ>50ИіАБЛљЫсЃЉЕФЛЏбЇКЯГЩШдШЛОпгаЬєеНадЃЌгШЦфЪЧдкДѓЙцФЃЩњВњжаЁЃЪЕбщЪвЙцФЃЕФыФКЯГЩЭљЭљНшжњЯжДњздЖЏыФКЯГЩвЧЃЈШчCEMLiberty PRIMEКЭCSBio IIЃЉздЖЏНјааЁЃетаЉаТаЭздЖЏыФКЯГЩвЧПЩвдЬсЙЉЖрДя192ИіВЛЭЌађСаЕФЫГађКЭЖрЦНааыФКЯГЩЃЌЪЙгУКьЭтЛђЮЂВЈМгШШвдЫѕЖЬЗДгІЪБМфЃЌгаЪБЪЙгУзЯЭтЯпМрВтвдШЗБЃЭбБЃЛЄЙ§ГЬЁЃДЫРрКЯГЩЦїЖдгкЪЕбщЪвЙцФЃЕФыФКЯГЩМЋЮЊгагУЃЌПЩвдПьЫйЩњВњЫљашЕФыФЃЌвдЙЉНјвЛВНЕФНсЙЙКЭЙІФмбаОПЁЃШЛЖјЃЌгЩгкШБЗІДѓаЭЩшБИКЭВЛОљдШЕФЙ§ШШЃЌКьЭтКЭЮЂВЈМгШШдкДѓЙцФЃыФжЦдьжаЕФгІгУгаЯоЃЌетПЩФмЕМжТИБВњЮяЕФВњЩњЁЃвђДЫЃЌДѓЖрЪ§СМКУЩњВњЙцЗЖ(GMP)ЧуЯђгкЮТКЭЕФЗДгІЬѕМўвдзюДѓЯоЖШЕиМѕЩйИБЗДгІКЭЯрЖддгжЪЃЌвђДЫДѓЙцФЃЩњВњГЄыФЃЈ>50ИіАБЛљЫсЃЉШдШЛОпгаЬєеНадЁЃ
ЛЏбЇыФКЯГЩММЪѕЕФЗЂеЙЃЌгШЦфЪЧSPPSММЪѕЕФЗЂеЙЃЌДѓДѓМгЫйСЫжЮСЦадыФЕФПЊЗЂЁЃвЛаЉжизщыФвЉЮяЃЌШчДпВњЫиКЭЬиСЂХСыФЃЌВЩгУЛЏбЇКЯГЩРДЩњВњЛюадвЉЮяГЩЗжЁЃыФЕФЛЏбЇКЯГЩЛЙдЪаэЖдЦфНјааЧЇБфЭђЛЏЕФаоЪЮЁЃ
ыФКЭыФФЃФтЮяЕФЛЏбЇаоЪЮ
ЖрыФзїЮЊвЛРрЬиЪтЕФжЮСЦвЉЮяЃЌЦфЩњЮяЛюадгыЦфЛЏбЇНсЙЙУмЧаЯрЙиЃЌЖрыФКЯГЩКѓашвЊРћгУвЉЮяЛЏбЇММЪѕНјаааоЪЮЃЌФЃФтЁЂЮШЖЈЛђЙЙНЈРэЯыЕФЖўМЖНсЙЙЃЌвдЬсИпЦфЩњЮяЛюадЃЌЪЕЯжЖрыФвЉЮяЕФбЁдёадЁЂЮШЖЈадКЭШмНтадЁЃ
дкаоЪЮЯШЕМыФКђбЁвЉЮяжЎЧАЃЌБиаыШЗЖЈОпгаЫљашЩњЮябЇЬиадЕФзюаЁЛюадађСаЁЃОЕфађСаЩЈУшЃЌГЦЮЊБћАБЫсЩЈУшЃЌЭЈГЃгУгкНЋУПИіВаЛљЬцЛЛЮЊБћАБЫсвдВњЩњвЛЯЕСаЯШЕМыФРрЫЦЮяЃЌвдШЗЖЈФФаЉЙиМќВаЛљИГгшЯШЕМыФЕФЩњЮяЛюадЃКЛюадНЕЕЭБэУїБЛЬцЛЛЕФВаЛљКмживЊЃЌЖјЛюадВЛЯджјНЕЕЭБэУїБЛЬцЛЛЕФВаЛљЪЧЖргрЕФЁЃШЛКѓЖдЯШЕМыФЕФПЩЬцЛЛВаЛљКЭ C ЖЫКЭ N ЖЫНјааНјвЛВНаоЪЮЃЌвдВњЩњзюжеЕФыФвЉЮяЁЃ
ыФЙЧМмаоЪЮ
ЙЧМмаоЪЮЕФжївЊдвђжЎвЛЪЧЬсИпыФЕФЕААзЫЎНтЮШЖЈадЁЃыФжаЕФЕААзЫЎНтЮЛЕуПЩвдЭЈЙ§ЮШЖЈадбаОПКЭДњаЛЮяВтЖЈРДЪЖБ№ЁЃЙЧМмаоЪЮАќРЈгУ D-АБЛљЫсШЁДњ L-АБЛљЫсЁЂВхШыМзЛљАБЛљЫсЁЂвдМАВєШы ІТ-АБЛљЫс140КЭРрыФ , НЋетаЉЗЧЬьШЛАБЛљЫсв§ШыыФађСаЃЌЬиБ№ЪЧдкЕААзЫЎНтЮЛЕуЃЌЪЧбгГЄыФРрвЉЮябЊНЌАыЫЅЦкЕФгааЇВпТдЁЃвЛИіГЩЙІЕФР§згЪЧШћРДМгбЙЫиЃЌЫќРДдДгкМгбЙЫиЃЌОпгаЯрЫЦЕФАаБъбЁдёадЃЌЕЋбЊНЌАыЫЅЦкИќГЄЁЃ
ыФЕФВрСДаоЪЮ
ыФЕФВрСДаоЪЮЪЧдкыФКЯГЩЙ§ГЬжагУЦфРрЫЦЮяЬцЛЛЬьШЛАБЛљЫсЃЌвдЬсИпЦфНсКЯЧзКЭСІКЭАаБъбЁдёадЁЃЬьШЛАБЛљЫсРрЫЦЮяЕФБфЬхЃЌШчИпОЋАБЫсЁЂмабѕРвАБЫсКЭ ІТ-БНБћАБЫсЃЌЭЈГЃЪЧЪаЪлЕФЃЌВЂЧвПЩвдЗНБуЕигУгкдкыФКЯГЩЙ§ГЬжаЖдыФВрСДНјааЛЏбЇаоЪЮЁЃМИжж GLP-1 РрЫЦЮявЉЮяЃЌШчРћРТГыФКЭЫїТэТГыФЃЌЖМгааоЪЮЕФВрСДЁЃ
ЭЈЙ§жїСДКЭВрСДаоЪЮФЃФтКЭЮШЖЈЖўМЖНсЙЙ
ыФжаЕФШѕСІЃЌШчЧтМќЁЂЗЖЕТЛЊСІКЭЗжзгФкЪшЫЎЯрЛЅзїгУВЛзувдаЮГЩЮШЖЈЕФЖўМЖНсЙЙЙЙЯѓЁЃвђДЫЃЌашвЊЖджїСДЁЂN ЖЫЛђ C ЖЫЛђВрСДНјааЖюЭтЕФаоЪЮЃЌвдФЃФтЬьШЛВњЮяЕФНсЙЙЛђ PPI жаЕФШШЕуЃЌВЂЮШЖЈЖўМЖНсЙЙЃЌвдВњЩњгаЧАЭОЕФыФРрвЉЮяКђбЁЮяЁЃ
ыФЛЗЛЏЁЃЛЗЛЏЪЧвЛжжГЃМћЕФыФаоЪЮММЪѕЃЌПЩвдАќРЈИїжжВпТдЃЌР§ШчЭЗЖдЮВЁЂжїСДЖдВрСДКЭВрСДЖдВрСДЛЗЛЏЃЈЭМ7ЃЉЁЃыФЛЗЛЏПЩвддіМгЕААзЫЎНтЮШЖЈадКЭЯИАћЭЈЭИадЃЌВЂПЩвдФЃФтКЭЮШЖЈыФЕФЖўМЖНсЙЙЁЃЕЅИіыФађСаШчЙћВЛгыЦфЫћыФСЌНгЃЌОЭВЛФмаЮГЩЛЗЛђзЊНЧНсЙЙЃЌЕЋЛЗЛЏЭЈЙ§дЄЯШзщжЏЗжзгФкЯрЛЅзїгУДйНјетаЉЖўМЖНсЙЙЕФаЮГЩЁЃыФЛЗЛЏвВГЃгУгкЮШЖЈЦфЫћЖўМЖНсЙЙЃЌШч ІС-Тна§КЭ ІТ-елЕўЁЃ
ыФФЃФтІС-Тна§КЭЮШЖЈЛЏЁЃТна§ЪЧзюГЃМћЕФЕААзжЪЖўМЖНсЙЙРраЭжЎвЛЃЌдМеМЫљгаЕААзжЪНсЙЙЕФ30%-40%ЁЃІС-Тна§гЩЗжзгФкЧтМќаЮГЩЃЌеМТна§НсЙЙЕФ90%ЁЃФЃФтыФжаЕФ ІС Тна§ПЩвдМјЖЈ PPI ЕФЕїНкМСЁЃПЩвдЭЈЙ§ВрСДНЈСЂНЛСЊЛђгУЙВМлМќШЁДњЧтМќЃЈГЦЮЊЧтМќЬцДњЮяЃЌHBSЃЉРДЮШЖЈ ІС Тна§ЁЃдк ІС Тна§НсЙЙжаЃЌЮЛжУ iЁЂi + 4 КЭ i +7 ЕФАБЛљЫсВрСДЮЛгкЭЌвЛВрЃЌЭЈЙ§ i КЭ i + 4 Лђ i КЭ i +7 НЈСЂНЛСЊПЩвдгааЇНгНќжїСДдзгВЂгажњгкдкТна§НсЙЙжааЮГЩЧтМќЁЃШЫУЧИЖГіСЫОоДѓХЌСІРДбаОПВЛЭЌЕФНЛСЊЃЌР§ШчЛљгкФкѕЃАЗЕФНЛСЊЃЈЭМ7)ЃЌЭЈЙ§ЙШАБЫсЛђЬьЖЌАБЫсгыРЕАБЫсВрСДаЮГЩФкѕЃАЗЧХЃЌЭЈЙ§гУАыызАБЫсЛђЭЌаЭАыызАБЫсЬцЛЛВаЛљаЮГЩЖўСђМќЃЌвдМАЫЋЧзЕчСЌНгзгЁЃЖЄКЯыФЪЧзюНќв§ШыЕФвЛжжаТЕФНЛСЊЗНЗЈЃЌгУгкЮШЖЈІС-Тна§НсЙЙЃЌЪЙгУЗЧЬьШЛЧзЕчАБЛљЫсЬцЛЛiКЭi + 4ЛђiКЭi +7ЮЛжУЕФВаЛљЃЌВЂгыЧзКЫНЛСЊаЮГЩСЌНгЁЃHBSаоЪЮВпТдАќРЈгУЙВМлМќШЁДњІС-Тна§ыФЕФвЛИіЧтМќЃЌвддЄЯШзщжЏТна§НсЙЙЁЃCabezasКЭSatterthwaitЪзЯШЪЙгУыТМќЙЙНЈHBSыФРДФЃФтІС-Тна§ЁЃAroraбаОПаЁзщвВЖдHBSыФНјааСЫЙуЗКЕФбаОПЃЌЪЙгУЯЉЬўСЌНгЬхРДЮШЖЈІС-Тна§ЁЃЫћУЧзюНќПЊЪМЪЙгУHBSВпТдРДЮШЖЈІТ-ЗЂМаЃЌвдМАетаЉаоЪЮыФЕФЩњЮяЛюадЃЌЮвУЧдкЧАЦкЙЄзїжавВдЫгУСЫHBSыФаоЪЮВпТдЃЌжиЕуЩшМЦСЫвЛИіЭъећЕФSPPSЭООЖЃЌвдМђЛЏHBSдкІСТна§ФЃФтКЭЮШЖЈжаЕФгІгУЁЃ
ІТ СДКЭ ІТ елЕўЕФыФФЃФтЁЃІТ елЕўКЭ ІТ СДДњБэСэвЛРрЛљгкзЊНЧФЃФтЕФЕААзжЪЖўМЖНсЙЙЁЃаоЪЮыФвдЮШЖЈ ІТ елЕўЭЈГЃЪЧЭЈЙ§в§Шы D-АБЛљЫсЃЈШч D-Pro ЃЉдкађСажааЮГЩзЊНЧНсЙЙРДЪЕЯжЕФЁЃD-Pro-L-Pro ФЃАхЪЧжкЫљжмжЊЕФжЇМмЃЌгУгкЮШЖЈМИжжГЩЙІЕФ PPI вжжЦМСжаЕФЗДЯђЦНаа ІТ ЗЂМа ЁЃДѓЛЗЛЏЛђЕэЗлбљЕААзІТ елЕўФЃФтЮявВвбгУгкДДНЈ ІТ елЕўКЭ ІТ СДНсЙЙЁЃ
ЛЏбЇаоЪЮЪЧжЦБИЫљашНсЙЙЕФЖрыФРрЫЦЮяЕФгааЇЗНЗЈЃЌЦфЮШЖЈадКЭЛюадЕФЬсИпвбЪЙЖржжЖрыФвЉЮяНјШыСйДВЃЌШчШќРжМгбЙЫиЁЂРћРТГыФЁЂЫїТэТГыФЕШЁЃШЛЖјгааЉаоЪЮВЂВЛФмЭЌЪБЬсИпЕААзЫЎНтЮШЖЈадКЭЛюадЃЌР§ШчВхШыD-АБЛљЫсЭЈГЃгажњгкбгГЄЖрыФЕФбЊНЌАыЫЅЦкЃЌЕЋD-АБЛљЫсаоЪЮЕФЖрыФКмЩйБэЯжГігааЇЕФЩњЮяЛюадЁЃ
жизщММЪѕЩњВњыФ
ЛЏбЇКЯГЩЪЧЙЄвЕжЦБИыФЕФЪзбЁЗНЗЈЃЌвђЮЊЫќПЩвдв§ШыГ§ЕААзжЪАБЛљЫсжЎЭтЕФЖрЙІФмКЯГЩНсЙЙЕЅдЊЃЌР§ШчЗЧЬьШЛАБЛљЫсКЭЩњЛЏЛђЩњЮяЮяРэЬНеыЃЌДгЖјдЪаэНјвЛВНаоЪЮЛђНсКЯЁЃДЫЭтЃЌЛЏбЇКЯГЩЙ§ГЬПЩвдЭъШЋздЖЏЛЏЃЌВЂЧввзгкРЉДѓЙцФЃЁЃЫќЮЊЩњВњЖЬыФКЭжаыФЬсЙЉСЫвЛжжЗНБугааЇЕФЗНЗЈЃЌЕЋГЄыФЕФЛЏбЇКЯГЩШдШЛОпгаЬєеНадЃЌвђДЫашвЊЬцДњВпТдЁЃ
Г§СЫЛЏбЇКЯГЩЭтЃЌжЮСЦадыФЛЙПЩвдЭЈЙ§ИїжжЩњЮяЗНЗЈжЦБИЃЌР§ШчЭЈЙ§ЬсШЁДгЬьШЛРДдДЗжРыЩњЮяЛюадыФЁЂУИКЯГЩЁЂЗЂНЭЁЂжизщ DNA ММЪѕКЭАыКЯГЩЁЃетаЉЗНЗЈПЩвдЕЅЖРгІгУЛђзщКЯгІгУЃЌОпЬхШЁОігкжЦБИыФЕФИДдгадКЭФбЖШЁЃ
ДгЬьШЛРДдДЗжРыыФРрвЉЮяЕФЪЕМљПЩвдзЗЫнЕН20ЪРМЭ20ФъДњЃЌЕБЪБвШЕКЫиЪзДЮДгЩќаѓвШЯйжаЗжРыГіРДВЂгУгкжЮСЦЬЧФђВЁЃЌЭьОШСЫЪ§ЪЎЭђШЫЕФЩњУќЁЃвШЕКЫиЕФПЊДДадГЩЙІЕМжТЙЋжкЖдыФСЦЗЈЕФШШЧщШевцИпеЧЃЌЫцКѓЦфЫћМИжжЖЏЮядДыФРрвЉЮяГЩЙІНјШыСйДВЪЙгУЃЌР§ШчДйЩіЩЯЯйЦЄжЪМЄЫиКЭНЕИЦЫиЗЧКЫЬЧЬхКЯГЩыФЪЧСэвЛИіживЊЕФЬьШЛРДдДМвзхЃЌПЩгУгкМјЖЈКЭЩњВњОпгажЮСЦЧБСІЕФыФЃЌР§ШчЭђЙХУЙЫиКЭЛЗцпОњЫиЁЃгыКЫЬЧЬхКЯГЩыФЛђЕААзжЪВЛЭЌЃЌЗЧКЫЬЧЬхКЯГЩыФЕФКЯГЩгЩБрТыЗЧКЫЬЧЬхыФКЯГЩУИЕФЛљвђДиПижЦЃЌЖјВЛЪЧФкдДадЗвыЛњжЦЃЌДгЖјВњЩњНсЙЙКЭЙІФмЖрбљЕФыФЃЌВЂЪЙетаЉЗжзгФмЙЛПЫЗўЦеЭЈыФвЉЮяЕФЙЬгаОжЯоадЁЃЖОвККЭЖОЫиБЛШЯЮЊЪЧМјЖЈЩњЮяЛюадыФЕФБІЙѓЬьШЛРДдДвдМАЦфЫћЬьШЛРДдДЃЌШчЛЗыФКЭыФСДыФвВЕУЕНСЫбаОПКЭРћгУЁЃУИКЯГЩЪЪгУгкКЯГЩЖЬыФЃЌШчЖўыФКЭШ§ыФЃЌУИКЯГЩыФвбГЩЙІгІгУгкЪГЦЗЬэМгМСЁЂвЉЮяКЭХЉгУЛЏбЇЦЗЕФЩњВњЁЃЗЂНЭвбБЛГфЗжжЄУїЪЧвЛжжЩњВњЩњЮяЛюадыФЕФЛЗБЃЗНЗЈЃЌР§ШчдкЛЗцпОњЫиЕФЩњВњжажизщDNAММЪѕФмЙЛЩњВњОпгаШЗЖЈађСаКЭЭЌжЪадЕФыФКЭЕААзжЪЁЃетжжЗНЗЈЖдгкжЦдьОпгаЖрИіЖўСђМќЕФГЄыФЛђИДдгыФЬиБ№гагУЃЌЗёдђетаЉыФКмФбЭЈЙ§ЛЏбЇКЯГЩЁЃШЫРрвШЕКЫиКЭЩњГЄМЄЫиЪЧЪЙгУжизщDNAММЪѕжЦдьЕФаэЖрПЩгУыФвЉЮяЕФДњБэадР§згЁЃДЫЭтЃЌжизщDNAММЪѕПЩвдгывХДЋУмТыРЉеЙКЭЦфЫћаТММЪѕЯрНсКЯЃЌЭЈЙ§ВєШыЗЧЬьШЛАБЛљЫсНЋЫљашЕФЙІФмЛљЭХв§ШыЗжзгжаЃЌШчЯТЫљЪіЁЃАыКЯГЩЬсЙЉСЫвЛжжСщЛюЕФЗНЗЈЃЌЭЈЙ§СЌНгКЯГЩыФКЭжизщDNAБэДяЕФыФРДЩњВњДѓаЭЩњЮяЛюадЖрыФЃЌЕБашвЊНјааЖрДЮШЫЙЄаоИФЪБЃЌетЪЧвЛжжЬиБ№гагУЕФЗНЗЈЁЃ
ЭЈЙ§вХДЋУмТыРЉеЙНјааЖрыФаоЪЮ
ЬьШЛЕААзжЪгЩ 20 жжЕфаЭАБЛљЫсКЯГЩЃЌетжжгаЯоЧвБЃЪиЕФАБЛљЫсПтЯджјЯожЦСЫЕААзжЪНсЙЙКЭЙІФмЕФЖрбљадКЭИДдгадЁЃвХДЋУмТыРЉеЙЪЧЖўЪЎФъЧАПЊЗЂЕФвЛжжММЪѕЃЌжМдкПЫЗўетвЛЯожЦЃЈЭМ8 ЃЉ ЁЃвХДЋУмТыРЉеЙдЪаэдкЕААзжЪЗвыЙ§ГЬжаНЋОпгааТЛЏбЇКЭЮяРэЬиадЕФЗЧЕфаЭАБЛљЫс ( ncAA ) ЮЛЕуЬивьадЕиВєШые§дкЩњГЄЕФЖрыФжаЃЌвЊЪЕЯжетвЛЕуашвЊЫФИізщГЩВПЗжЃК1ЃЉОпгаЫљашЛЏбЇКЭЮяРэЬиадЕФ ncAAЃЛ2ЃЉжИЖЈ ncAA ЕФЖРЬиУмТызгЃЌР§ШччњчъжежЙУмТызг (UAG) ЛђЫФСЊЬхУмТызгЃЛ3ЃЉвжжЦЖРЬиУмТызгЧвВЛгыЦфФкдДЖдгІЮяЗЂЩњДЎШХЕФе§НЛ tRNAЃЛ4ЃЉПЩвдНЋ ncAA ЬивьадЕиГфЕчЕНе§НЛ tRNA ЩЯЧвВЛгыФкдДадАБЛљѕЃЛљ tRNA КЯГЩУИ/tRNA Жде§НЛАБЛљѕЃЛљtRNAКЯГЩУИЗЂЩњДЎШХЁЃ
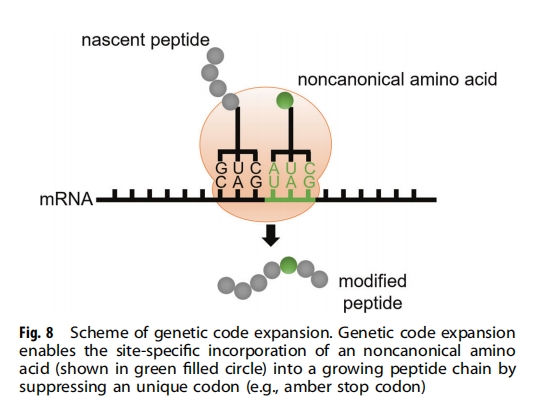
ЦљНёЮЊжЙЃЌвбга 200 ЖржжОпгаВЛЭЌЙІФмЕФ ncAA БЛЛљвђБрТыЕНВЛЭЌЕФЩњЮяЬхжаЃЌР§ШчДѓГІИЫОњЁЂНЭФИЁЂВИШщЖЏЮяЯИАћЁЂВЁЖОЩѕжСЖЏЮяЃЌЮЊЕААзжЪбаОПКЭЙЄГЬЬсЙЉСЫБІЙѓЕФЙЄОпЯфЁЃетЬзРЉеЙЕФЙЙНЈФЃПщАќРЈЩњЮяе§НЛЛЏбЇНсКЯАщТТЁЂН№ЪєђќКЯМСЁЂЙтНЛСЊМСЁЂСкНќНЛСЊМСЁЂЙтС§БЮАБЛљЫсЁЂОЙ§ЗвыКѓаоЪЮЕФАБЛљЫсЃЈСзЫсЛЏЁЂСђЫсЛЏЁЂѕЃЛЏЕШЃЉЁЂбѕЛЏЛЙдЛюадАБЛљЫсвдМАКьЭтЁЂКЫДХЙВеёЁЂгЋЙтЬНеыЃЌвбЙуЗКгІгУгкЕААзжЪЕФбаОПЁЂВйзїКЭНјЛЏЁЃвХДЋБрТыЖржж ncAA ЕФФмСІдЪаэКЯРэгХЛЏКЭЩњВњОпгаУїШЗНсЙЙЁЂЙІФмКЭЛЏбЇМЦСПЕФЛЏбЇаоЪЮжизщЕААзЁЃдкетРяЃЌЮвУЧжиЕуЙизЂвХДЋУмТыРЉеЙдкжЮСЦадыФКЭЕААзжЪНјЛЏжаЕФгІгУЁЃ
ыФКЭЕААзжЪЕФОлввЖўДМЛЏ
ЭЈЙ§вХДЋУмТыРЉеЙВњЩњЕФЖЬЕААзКЭыФжЮСЦМСгЩгкЦфвЉДњЖЏСІбЇНЯВюЃЈАќРЈПьЫйбЊЧхНЕНтКЭПьЫйЯћГ§ЃЉЃЌАыЫЅЦквВНЯЖЬЁЃСЌНгОлКЯЮяЪЧбгГЄЕААзжЮСЦМСАыЫЅЦкЕФвЛжжЗНЗЈЁЃPEG гЩжиИДЕФЛЗбѕввЭщЕЅдЊаЮГЩЃЌЪЧвЛжжВЛПЩЩњЮяНЕНтЁЂЮоЖОЁЂЕЭУтвпдадЕФОлКЯЮяЁЃPEG ЛЏПЩвддіМгЕААзжЪЕФгааЇЗжзгСПЃЌДгЖјМѕЩйЩідрЙ§ТЫЕФЩідрЧхГ§ТЪЁЃPEG ВПЗжЛЙПЩвдЭЈЙ§діМгПеМфЮЛзшРДБЃЛЄЕААзжЪВЛБЛЕААзЫЎНтУИЯћЛЏЃЌВЂЭЈЙ§діМгФПБъЕААзжЪЕФЫЎШмадРДАяжњдіМгЮќЪеЃЌетаЉгХЪЦЪЙЕУОлввЖўДМЛЏГЩЮЊаоЪЮжЮСЦадЕААзжЪЕФЦеБщВпТдЃЌзд 1970 ФъДњвдРДЃЌОлввЖўДМЛЏвбгІгУгкгХЛЏЕААзжЪжЮСЦЃЌВЂШЁЕУСЫОоДѓГЩЙІЁЃФПЧАЪаГЁЩЯга 10 ЖржжОлввЖўДМЛЏЕААзжЪжЮСЦМСЃЌЛЙгаИќЖрЧБдкКђбЁвЉЮяе§дкСйДВЪдбщжаЁЃ
ДЋЭГЕФОлввЖўДМЛЏЭЈГЃЗЂЩњдкРЕАБЫсЛђАыызАБЫсВаЛљЁЃШЛЖјЃЌШчЙћФПБъЕААзжЪАќКЌЖрИіЗДгІадРЕАБЫсЛђАыызАБЫсВаЛљЃЌгЩгкШБЗІбЁдёадЃЌНсКЯПЩФмЫцЛњЗЂЩњдкетаЉВаЛљжаЕФШЮКЮвЛИіЩЯЃЌДгЖјВњЩњФбвдЗжРыЕФвьжЪНсКЯВњЮяЁЃвђДЫашвЊдЪаэЮЛЕуЬивьадОлввЖўДМЛЏЕФММЪѕЃЌЦфжаОлввЖўДМВПЗжПЩвдбЁдёадЕиКЭЮЛжУПижЦЕиСЌНгЕНЕААзжЪЩЯЃЈЭМ9ЃЉЁЃ
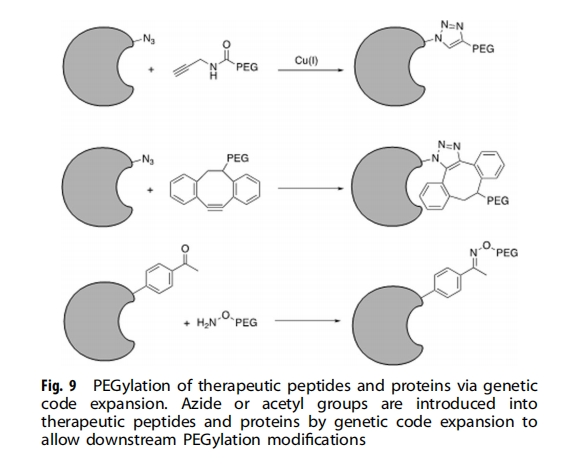
вХДЋУмТыРЉеЙЮЊЕААзжЪОлввЖўДМЛЏЬсЙЉСЫБІЙѓЕФЙЄОпЁЃвЛжжЗНЗЈЪЧНЋКЌгаЩњЮяе§НЛЛЏбЇЪжБњЕФ ncAAЃЈЭМ10ЃЉвХДЋБрТыЕНФПБъЕААзжЪжаЃЌШЛКѓЭЈЙ§ЩњЮяе§НЛЗДгІгы PEG НсКЯЁЃ2004 ФъЃЌDeiters ЕШШЫБЈЕРСЫЕквЛжжЛљгкНЭФИжаЖдЕўЕЊБНБћАБЫс (pAzF) ЕФвХДЋећКЯЕФ ncAA НщЕМЕФЕЅОлввЖўДМЛЏЗНЗЈЁЃЭЈЙ§Э (I) ДпЛЏЕФШВЬў-ЕўЕЊЛЏЮяЛЗМгГЩ (CuAAC) ЕуЛїЗДгІНЋШВЬўбмЩњЕФ PEG СДЮЛЕуЬивьадЕиСЌНгЕНГЌбѕЛЏЮяЦчЛЏУИ (SOD) ЩЯЃЌЫљЕУ SOD ЯдЪОГігывАЩњаЭЕААзжЪЯрЫЦЕФУИЛюадЁЃеХЕШШЫБэУїетжжЕЅОлввЖўДМЛЏЗНЗЈвВПЩвдгІгУгкИЩШХЫи (IFN)-ІС2bЁЃНЋДјгаЕўЕЊЛЏЮяЕФ ncAA NEAK ЮЛЕуЬивьадЕиећКЯЕН IFN-ІС2b ЕФВЛЭЌЮЛжУЃЌДгЖјЭЈЙ§ЮоЭЛЗМгГЩЗДгІгы PEG Нјаае§НЛКЭЛЏбЇМЦСПНсКЯЁЃдкФіГнЖЏЮяФЃаЭжаЃЌШ§жжЫљЕУ IFN-ІС2b БфЬхЯдЪОГіБШЦфЫћБфЬхКЭвАЩњаЭЗжзгИќИпЕФЩњЮяЛюадКЭИќКУЕФвЉДњЖЏСІбЇЬиеїЁЃЩЯЪіР§згБэУїЃЌЕЅОлввЖўДМЛЏПЩвдЦеБщЪЪгУгкИїжжЕААзжЪЁЃ
2011 ФъЃЌCho ЕШШЫБЈЕРСЫЭЈЙ§ЛљвђУмТыРЉеЙВњЩњЕФжизщЕААзЕФЪзДЮСйДВЪдбщЁЃЭЈЙ§дкВЛЭЌЮЛжУМгШыЖдввѕЃБНБћАБЫс (pAcF)ЃЌШЛКѓгы PEG НјааЮЛЕуЬивьадНсКЯЃЌжЦГЩСЫ 20 жжШЫРрЩњГЄМЄЫи (hGH) БфЬхЁЃвЛжждкВаЛљ 35 ДІНјааЕЅОлввЖўДМЛЏЕФ hGH БфЬхЃЌГЦЮЊ ARX201ЃЌдк GH ШБЗІЕФДѓЪѓжаБэЯжГіСМКУЕФвЉаЇбЇЬиадЃЌВЂЧвдкГЩФъ GH ШБЗІЛМепжаОпгагыЬьШЛ hGH СЦЗЈЯрЕБЕФСЦаЇКЭАВШЋадЃЌЕЋаЇСІдіЧПЧвзЂЩфЦЕТЪНЕЕЭЁЃWu ЕШШЫЫцКѓЃЌЭЈЙ§дк 35ЁЂ131 КЭ 145 ЮЛЕуДІЖдЖрИі NEAK НјаавХДЋБрТыЃЌЩњГЩСЫдкВаЛљ 35ЁЂ131 КЭ 145 ДІНјаазщКЯ PEG ЛЏЕФ hGH БфЬхЁЃгыЕЅ PEG ЛЏ hGH ЯрБШЃЌЫљЕУЕФЖр PEG ЛЏ hGH БфЬхБэЯжГіИќЕЭЕФУтвпдадКЭИќКУЕФвЉДњЖЏСІбЇЬиадЃЌЧвВЛЫ№ЪЇЩњЮяЛюадЃЌВЂЧвдкФіГнЖЏЮяФЃаЭжаБШЕЅ PEG ЛЏ hGH ОпгаИќИпЕФЮШЖЈадЁЃетаЉЪОР§ЫЕУїСЫвХДЋУмТыРЉеЙЖдгкгХЛЏжЮСЦадЕААзжЪКЭыФЕФЪЕгУадЁЃ
СэвЛжжЮЛЕуЬивьадОлввЖўДМЛЏЗНЗЈЪЧЭЈЙ§вХДЋУмТыРЉеЙНЋКЌ PEG ЕФ ncAA жБНгв§ШыАаЕААзЁЃShozen ЕШШЫЪЙгУЮоЯИАћЗвыЯЕЭГвжжЦЫФСЊЬхУмТызгЃЌЮЛЕуЬивьадЕиВєШыСЫКЌга PEG4ЁЂPEG8 КЭ PEG12 СДЕФ ncAAЁЃTada ЕШШЫЪЙгУРрЫЦЕФВпТдЃЌЭЈЙ§вжжЦчњчъжежЙУмТызгЃЌНЋ PEG4 жС PEG24 ЕФНЯГЄ PEG СДв§ШыЖрыФЁЃFu ЕШШЫзюНќНЋ eN-И§ѕЃ-l-РЕАБЫс (HepoK) в§Шы GLP-1ЃЈЭМ10ЃЉ), ЫљЕУGLP-1(HepoK)гывАЩњаЭGLP-1ЯрБШ, ЖдШЫбЊЧхАзЕААз(HSA)ЕФНсКЯЧзКЭСІИќЧП, ЧвНЕбЊЬЧзїгУГжајЪБМфИќГЄ, ЮЊбаОПЕААзжЪжЌЛЏЬсЙЉСЫгаСІЕФЙЄОпЁЃ
вХДЋУмТыРЉеЙКЭ ncAA вВвбгУгкЩњГЩВЛЭЌРраЭЕФвпУчЃЌАќРЈыФвпУчЃЌШчбЧЕЅЮЛвпУчЁЂНсКЯвпУчКЭМѕЖОЛювпУчЁЃGrunewaldЕШШЫЪзДЮжЄУїНЋУтвпдад ncAA ећКЯЕНФПБъЕААзжаПЩвдДђЦЦздЩэЕААзЕФУтвпФЭЪмадВЂдкЖЏЮяФЃаЭжав§ЗЂУтвпЗДгІЁЃОпЬхЖјбдЃЌНЋЖдЯѕЛљБНБћАБЫс (pNO2F) ЛђБНБћАБЫсЕФЕЅвЛЭЛБфв§ШыаЁЪѓжзСіЛЕЫРвђзг-ІС (mTNF-ІС) ЕФЕк 86 ЮЛЃЌЗжБ№ВњЩњ mTNF-ІС (pNO2F) КЭ mTNF-ІС (Phe)ЁЃЫљЕУЕФ mTNF-ІС (pNO2F) дкаЁЪѓжагеЕМИпЕЮЖШПЙЬхЗДгІЃЌЖј mTNF-ІС (Phe) дђВЛЛсЁЃДЫЭтЃЌЗЂЯж mTNF-ІС (pNO2F) геЕМЕФПЙЬхгыЬьШЛ mTNF-ІС ИпЖШНЛВцЗДгІЃЌВЂБЃЛЄаЁЪѓУтЪмжЌЖрЬЧ (LPS) геЕМЕФЫРЭіЁЃдкЫцКѓЕФЛњжЦбаОПжаЃЌGrunewald ЕШШЫЗЂЯж mTNF-ІС (pNO2F) ЭЛБфЬхПЩВњЩњ T ЯИАћвРРЕадЖрПЫТЁПЙЬхКЭПЙ mTNF-ІС IgG ПЙЬхЗДгІЃЌетжжЗДгІжСЩйГжај 40 жмЃЌВЂБЃЛЄаЁЪѓУтЪм LPS ДЬМЄв§Ц№ЕФбЯжиФкЖОбЊжЂЁЃетжжЗНЗЈЛЙв§ЗЂСЫЖдЪѓЪгЛЦДМНсКЯЕААзЕФИпЕЮЖШ IgG ПЙЬхЗДгІЃЌетБэУїетПЩФмЪЧНЋЦфЫћШѕУтвпдадздЩэЕААззЊЛЏЮЊвпУчЕФЦеБщЪЪгУЗНЗЈЁЃдкКѓајЪЕбщГ§ ncAAЃЈpNO2FЃЉЭтЃЌдкздЩэЕААзЕФЬиЖЈЮЛжУВхШыЬхЯИАћЭЛБфЃЈmTNF-ІС жаЕФ Tyr КЭ EGF жаЕФ PheЃЉКЭЗвыКѓаоЪЮЃЈ3NO2Tyr КЭ SO3TyrЃЉвВЛсв§Ц№еыЖдЬьШЛЕААзЕФЧПСв IgG ПЙЬхЗДгІЁЃЩЯЪіНсЙћБэУїЃЌЮЛЕуЬивьадВхШыУтвпдад ncAA КЭФГаЉЬьШЛЗвыКѓаоЪЮ (PTM) ПЩвдДђЦЦздЩэЕААзЕФУтвпФЭЪмадВЂВњЩњжЮСЦадвпУчЁЃ
Wang ЕШШЫНЋЖрИіОпгаБНБћАБЫсЙЧМмЕФ ncAA ећКЯЕННсКЫЗжжІИЫОњЁЂПЈНщУчКЭНсКЫЗжжІИЫОњжаЃЌвдДйНјНсКЫВЁвпУчЕФбаОПКЭПЊЗЂЁЃгЩгкВИШщЖЏЮяВЁЖОЕФДрШѕадКЭИДдгЕФзщзАЙ§ГЬЃЌЪЙгУГЃЙцЛЏбЇаоЪЮЗНЗЈКмФбВйзнЭъећЕФЛюВЁЖОЁЃЮЊСЫПЫЗўетвЛЬєеНЃЌLin ЕШШЫБЈЕРСЫЕквЛИіР§згЃЌНЋ ncAAs ЮЛЕуЬивьадЕиВєШыЭъећКЭЛюВЁЖОжаЃЌШЛКѓНјаабЁдёадБъМЧЃЌЖјВЛЛсЩЅЪЇДЋШОадЁЃОпЬхРДЫЕЃЌвЛзщпСПЉРЕАБЫсРрЫЦЮяБЛвХДЋБрТыЕНвваЭИЮбзВЁЖО (HBV) ЕФАќФЄЕААзжаЃЌВЂдкШЫИЮЯИАћжазщзАГЩЛюЖЁаЭИЮбзВЁЖО (HDV)ЃЌОпгабЯИёЕФбЁдёадКЭИпаЇТЪЁЃЭЈЙ§ЩИбЁВЛЭЌЕФВєШыЮЛЕуЃЌВЁЖОЕФДЋШОадЕУвдЭъШЋБЃГжЁЃДЫЭтЃЌncAA аоЪЮЕФВЁЖОПЩвдЭЈЙ§Э (I) ДпЛЏЕФШВЬў-ЕўЕЊЛЏЮяЛЗМгГЩЕуЛїЗДгІЧсЫЩЕиБЛРЯТЛђНсКЯЁЃWang ЕШШЫЛЙРћгУ ncAA НщЕМЕФЛљвђПЊЙиПЊЗЂСЫМѕЖОЛю HIV-1 впУчЁЃвЛзщБНБћАБЫсРрЫЦЮяБЛЛљвђБрТыЕН HIV-1 ЕФБиашЕААзжЪжавдПижЦЦфИДжЦЃЌЭЈЙ§етжжЗНЗЈПЩвдОЋШЗЕиДђПЊКЭЙиБе HIV-1 ИДжЦЁЃдкКѓајбаОПжаЃЌдЌЕШШЫНЋ ncAA НщЕМЕФЛљвђПЊЙиКЯВЂЕНВЁЖОЛљвђзщжаЃЌВЂПЊЗЂСЫЛљгкчњчъвжжЦЕФЖржмЦкПЩИДжЦ HIV-1ЃЌДњБэСЫ HIV-1 впУчПЊЗЂЕФживЊвЛВНЁЃГТЕШШЫЭЈЙ§вжжЦЬьШЛЕААзжЪЗвыЯЕЭГВЛЪЙгУЕФЫФСЊУмТызгЃЌЪЕЯжСЫЖд HIV-1 ИДжЦЕФОЋШЗПижЦЃЌДгЖјзюДѓЯоЖШЕиМѕЩйСЫаЃЖдЕФПЩФмадВЂЬсИпСЫвпУчЕФАВШЋадЁЃИУЗНЗЈЛЙгІгУгкМзаЭСїИаВЁЖОЃЌВЂВњЩњСЫАВШЋгааЇЕФМѕЖОЛювпУчЃЌетаЉвпУчдкЖЏЮяФЃаЭжав§ЗЂСЫЧПДѓЕФБЃЛЄадУтвпЗДгІЃЌетБэУї ncAA НщЕМЕФМѕЖОЛювпУчЪЧвЛжжЦеБщЪЪгУЕФЗНЗЈЁЃ
ЙВМлыФ/ЕААзжЪвЉЮя
аЁЗжзгЙВМлвЉЮягыЗЧЙВМлвЉЮяЯрБШОпгааэЖргХЪЦЃЌР§ШчЩњЛЏаЇТЪКЭаЇСІЬсИпЁЂвЉДњЖЏСІбЇИФЩЦЁЂзїгУЪБМфбгГЄЁЂМССПКЭИјвЉЦЕТЪМѕЩйвдМАЖдФбжЮадАаЕугаЧПаЇвжжЦзїгУЁЃгЩгкЦфбЁдёадЕЭКЭЙВМлвЉЮя-ЕААзжЪМгКЯЮяЕФЧБдкУтвпдадЕШАВШЋЮЪЬтЃЌШЫУЧгавтБмУтПЊЗЂаЁЗжзгЙВМлвЉЮяЁЃШЛЖјЃЌЛљгкЛюадЕФЕААзжЪЗжЮіКЭЦфЫћаТММЪѕЕФЗЂеЙвтЮЖзХаЁЗжзгЙВМлвЉЮяжиаТЪмЕНЙизЂЃЌМИжжЭЈЙ§ЙВМлНсКЯЛњжЦЦ№зїгУЕФаЁЗжзгвЉЮявбЛёХњЩЯЪаЁЃ
ДгРэТлЩЯНВЃЌЙВМлЕААзвЉЮягІИУОпгагыаЁЗжзгвЉЮяЯрЫЦЕФгХЪЦЁЃШЛЖјЃЌгЩгкЫќУЧЙЬгаЕФЮоЗЈаЮГЩЬьШЛЕААзжЪЕФЙВМлМќЃЌвђДЫЙВМлЕААзвЉЮяЕФжЮСЦЧБСІЩаЮДЕУЕНГфЗжЕФЬНЫїЁЃLiЕШШЫзюНќБЈЕРСЫвЛжжНгНќМЄЛюЗДгІСЦЗЈ(PERx)ВпТдРДПЊЗЂЙВМлЕААзвЉЮяЁЃЫћУЧНЋЧБдкЕФЩњЮяЛюадncAAЗњЛЧЫс- L -РвАБЫс(FSY)268вХДЋЕиНсКЯЕНШЫГЬађадЯИАћЫРЭіЕААз1(PD-1)ЕФ129ЮЛЃЌВЂБэУїЫљЕУЕНЕФPD-1(FSY)дкЬхЭтКЭЬхФкбЁдёадЕигыЦфЬьШЛХфЬхPD-L1аЮГЩЙВМлМќЁЃв§ШЫзЂФПЕФЪЧЃЌгывАЩњаЭPD-1ЯрБШЃЌPD-1(FSY)ЯджјдіЧПСЫШЫnaïve TЯИАћКЭЙЄГЬЧЖКЯПЙдЪмЬхTЯИАћЕФЩњЮяЛюадЁЃдкМИжжУтвпШЫдДЛЏаЁЪѓФЃаЭжаЃЌPD-1(FSY)ЯдЪОГіИќгааЇЕФжзСіЩњГЄвжжЦзїгУЃЌВЂОпгагыжЮСЦадПЙPD-L1ПЙЬхЯрЕБЛђИќДѓЕФПЙжзСізїгУЁЃШЛКѓЃЌЫћУЧНЋPERxгІгУгкfsyаоЪЮЕФеГИНЬхЖдHER2ЪмЬхЕФЙВМлвжжЦЃЌЫЕУїPERxПЩвдЮЊПЊЗЂЙВМлЕААзвЉЮяЬсЙЉвЛИіЭЈгУЦНЬЈЁЃгыЗЧЙВМлЕААзвЉЮяЯрБШЃЌPERxвЉЮяПЩвдвдЦфдЪМаЮЪНЪЙгУЃЌВЛашвЊЖюЭтЕФаоЪЮРДбгГЄЦфАыЫЅЦкЃЌвђЮЊЙВМлНсКЯЪЙвЉЮявЉаЇгыЦфвЉДњЖЏСІбЇНтёюЁЃДЫЭтЃЌPERxдЪаэPD-1 (15.6 kDa)ЕШаЁЕААзЩњЮяжЦМСгУзїжЮСЦвЉЮяЃЌДгЖјДѓДѓРЉеЙСЫжЮСЦЕААзЕФЗЖЮЇЁЃДЫЭтЃЌгЩгкЕААзжЪвЉЮягыЦфАаБъжЎМфЕФФкдкЧзКЭСІЃЌвдМАЧБдкЩњЮяЛюадncAAЕФСкНќЧ§ЖЏНЛСЊЛњжЦЃЌPERxПЩвдзюДѓЯоЖШЕиМѕЩйЭбАааЇгІЁЃетаЉгХЪЦвтЮЖзХPERxВпТдгаЧБСІЮЊПЊЗЂаТаЭЙВМлЕААзвЉЮяЬсЙЉвЛИіЭЈгУЦНЬЈЁЃPERxВпТдБГКѓЕФЛЏбЇКЭИќЖрЕФЙВМлЕААзЕФР§згвбОдкЦфЫћЕиЗНЯъЯИЛиЙЫ.
жЌжЪКЭНЯДѓЕФЕААзжЪОГЃБЛХМСЊвдИФЩЦЙВМлыФвЉЮяЕФвЉДњЖЏСІбЇЁЃвбЛёХњзМЕФыФвЉЮяЃЌШчРћРТГыФЁЂЫїТэТГыФКЭЕТЙШвШЕКЫиЃЌгы C 14/16/18 жЌЗОЫсНсКЯЃЌетдіМгСЫЫќУЧЕФбЊНЌбЛЗЪБМфВЂМѕЩйСЫЫќУЧдкЩідрЯћГ§Й§ГЬжаЕФНЕНтЁЃСНжжбЊНЌЕААзЃЌбЊЧхАзЕААзКЭУтвпЧђЕААзЃЌвВгУгкЭЈЙ§діМгЦфЗжзгСПРДбгГЄыФбЛЗЪБМфЃЌДгЖјГЌЙ§ЩіаЁЧђТЫЙ§ЕФЗжзгСПНижЙжЕЁЃР§ШчЃЌетжжВпТдБЛгУгкбгГЄЖШРТГыФКЭАЂБиТГыФЕФАыЫЅЦкЃЌУПжмзЂЩфвЛДЮЁЃ
ыФвЉЮяЪфЫЭЕФЗЂеЙ
ыФаоЪЮПЩЪЙыФЛёЕУИќКУЕФЛюадКЭбЊНЌЮШЖЈадЃЌВЂБфЕУИќЯёвЉЮяЁЃШЛЖјЃЌыФЕФЙЬгаЬиадвтЮЖзХЫќУЧКмШнвзБЛЮИГІЕРжаЕФЯћЛЏУИЫЎНтЃЌвђДЫДѓЖрЪ§ыФРрвЉЮяЪЧЭЈЙ§зЂЩфИјвЉЕФЁЃзюНќЕФбаОПвбОЬНЫїСЫыФРрвЉЮяЕФИјвЉЭООЖвдПЫЗўетаЉШБЕуЁЃ
гыЩјЭИДйНјМССЊКЯЪЙгУЪЧвЛжжКмгаЧАОАЕФВпТдЃЌПЩЪЕЯжыФРрвЉЮяЕФПкЗўИјвЉЁЃгы C18жЌЗОЫсНсКЯЕФЫїТэТГыФвбЛёзМЭЈЙ§УПжмвЛДЮЦЄЯТзЂЩфИјвЉ, ЦфбЊНЌЮШЖЈадгХгкЦфЫћGLP-1РрЫЦЮяЁЃИќСюШЫЙФЮшЕФЪЧЃЌЫїТэТГыФгыN-[8-(2-єЧЛљБНМзѕЃАБЛљ]аСЫсФЦ (SNAC) ЕФИДКЯжЦМСвбЛёХњгУгкПкЗўжЮСЦ 2 аЭЬЧФђВЁЁЃгы SNAC ЕФИДКЯжЦМСПЩНЕЕЭЯћЛЏУИЕФЙІаЇЃЌДгЖјЗРжЙЫїТэТГыФдкЮИжаБЛЦЦЛЕЁЃЪшЫЎад SNAC ЗжзгЛЙдіМгСЫЫїТэТГыФЕФЧзжЌадЃЌДгЖјИФЩЦЦфЭЈЙ§ЮИФЄЕФПчЯИАћЮќЪеМАЦфНјШыЬхбЛЗЕФзЊдЫЁЃгыЦфЫћЩјЭИДйНјМСЁЂУИвжжЦМСКЭЫЎФ§НКЕФИДКЯжЦМСвВвбгУгкдЪаэПкЗўЦфЫћыФРрвЉЮяЃЌШчАТЧњыФКЭвШЕКЫиЃЌетаЉвЉЮяФПЧАе§дкСйДВЪдбщжаЁЃФПЧАе§дкбаОПИќЖрВпТдЃЌАќРЈЗЮВПИјвЉЁЂЭИЦЄИјвЉКЭЪЙгУжВШыЪНБУЃЌгУгкЪфЫЭЬиЖЈыФРрвЉЮяЃЌАќРЈПЊЗЂЮќШыЪНвШЕКЫиКЭгУгквШЕКЫиЪфЫЭЕФЮЂаЭжВШыЪНБУЁЃЮвУЧдЄМЦетаЉММЪѕНЋдкЮДРДМИФъгІгУгкИќЖрыФРрвЉЮяЁЃ
жЮСЦадыФдкМВВЁСьгђЕФЗЂеЙМАгІгУЯжзД
жЮСЦадыФдкЬЧФђВЁжЮСЦжаЕФгІгУ
2 аЭЬЧФђВЁЪЧгЩКѓЬьвШЕКЫиШБЗІв§Ц№ЕФЃЌдкжаРЯФъШЫжаКмГЃМћЁЃ2 аЭЬЧФђВЁвбГЩЙІЭЈЙ§ыФРрвЉЮяжЮСЦЃЌАќРЈ GLP-1 ЪмЬхМЄЖЏМС (GLP-1RA) КЭзюжјУћЕФыФРрвЉЮявШЕКЫиЁЃGLP-1 ЪЧвЛжжгЩЛиГІ L ЯИАћЗжУкЕФФкдДадЩњГЄМЄЫиЁЃЦфЪмЬхДцдкгквШЯй ІТ ЯИАћЁЂЭтжмКЭжаЪрЩёОЯЕЭГЁЂаФдрКЭбЊЙмЁЂЩідрЁЂЗЮКЭЮИГІЕРеГФЄжаЃЈЭМ11ЃЉЁЃGLP-1 гыЦфЪмЬхЯрЛЅзїгУЃЌДЬМЄвШЕК ІТ ЯИАћЗжУквШЕКЫиЃЌвжжЦвШЕК ІС ЯИАћЪЭЗХвШИпбЊЬЧЫиЃЌдіМгБЅИЙИаЃЌВЂвдЦЯЬбЬЧвРРЕЕФЗНЪНбгЛКЮИХХПеЁЃФкдДадGLP-1БЛЖўыФЛљыФУИ-4ЃЈDPP-4ЃЉНЕНтВЂбИЫйЪЇЛюЁЃЮЊСЫбгГЄGLP-1ЪмЬхЕФДЬМЄЪБМфЃЌашКЯГЩGLP-1RAsРДзшжЙЦфНЕНтЁЃзд2005ФъЕквЛИіGLP-1RAЁЊЁЊАЌШћФЧыФБЛУРЙњЪГЦЗвЉЦЗМрЖНЙмРэОжЃЈFDAЃЉХњзМвдРДЃЌЖрИіGLP-1RAsНјШыСйДВЃЌАќРЈРћРТГыФЃЈ2009ФъЃЉЁЂРћЮїРРДыФЃЈ2013ФъЃЉЁЂЖШРТГыФЃЈ2014ФъЃЉЁЂЫїТэТГыФЃЈ2017ФъЃЉЁЃетаЉGLP-1RAsзЂЩфКѓФмгааЇНЕЕЭЬЧЛЏбЊКьЕААзКЭЦНОљбЊЬЧЫЎЦНЃЌИФЩЦПеИЙбЊЬЧЁЃ
вЛаЉGLP-1RAsЖджЮСЦФГаЉ2аЭЬЧФђВЁВЂЗЂжЂвВгааЇЁЃЬЧФђВЁЩіВЁЪЧ2аЭЬЧФђВЁзюЮЃЯеЕФВЂЗЂжЂжЎвЛЃЌЖдЬЧФђВЁЛМепЕФЩіЙІФмдьГЩбЯжигАЯьЃЌСйДВБэЯжАќРЈЕААзФђКЭЩіаЁЧђТЫЙ§ТЪЃЈGFRЃЉЯТНЕЁЃдквЛЯюЖд35Ућ2аЭЬЧФђВЁЛМепЕФбаОПжаЃЌРћРТГыФЭЈЙ§вжжЦНќЖЫаЁЙмФЦЧтЗДЯђзЊдЫЕААз3ЃЈNHE3ЃЉДгЖјдіМгФЦЁЂТШКЭМиЕФОјЖдКЭВПЗжХХаЙЃЌВЂдіМгФђвКpHжЕЃЌДгЖјНЕЕЭСЫУОЁЂИЦКЭСзЫсбЮЕФОјЖдКЭВПЗжХХаЙЃЌгыИЪОЋвШЕКЫиЯрБШЁЃДЫЭтЃЌдквЛЯюЖд30Ућ2аЭЬЧФђВЁЛМепЕФбаОПжаЃЌРћРТГыФЯджјНЕЕЭСЫGFRЁЂФђАзЕААзХХаЙТЪКЭВПЗжАзЕААзХХаЙ . GLP-1RAs ПЩЭЈЙ§діМгФЦЯђжТУмАпЕФСїГіЁЂдіМгЩіаЁЙм-ЩіаЁЧђЗДРЁКЭДЋШыаЁЖЏТіЕФбЊЙмЪеЫѕРДНЕЕЭ GFRЃЌВЂЧвЛЙПЩФмЭЈЙ§НЕЕЭбЊНЌЩіЫиЛюадЁЂНЕЕЭЩідрбѕЛЏгІМЄКЭдіМгХХФЦРДМѕЩйАзЕААзФђЁЃШЛЖјЃЌетаЉгАЯьдкЖрДѓГЬЖШЩЯЪЧгЩ GLP-1R НщЕМЕФШдгаД§ШЗЖЈЁЃзюНќЕФбаОПжЄЪЕЃЌGLP-1 ЕФДњаЛЮяБЃСєСЫживЊЕФПЙбѕЛЏКЭПЙЕђЭіЛюадЃЌетаЉЛюадгы GLP-1R ЮоЙиЁЃаФбЊЙмМВВЁШдЪЧ2аЭЬЧФђВЁЛМепЫРЭіЕФжївЊдвђЃЌвђДЫдкбЁдё2аЭЬЧФђВЁжЮСЦЗНАИЪБгІПМТЧаФбЊЙмВЂЗЂжЂЕФЗРжЮЁЃGLP-1RAsвбБЛжЄЪЕЖдаФбЊЙмМВВЁгагавцзїгУЁЃзюНќЕФСйДВЪдбщЗЂЯжЃЌжЛгаРћРТГыФКЭЫїТэТГыФдкаФбЊЙмвцДІЗНУцгагХЪЦЃЌОЁЙмЦфЛњжЦЩаВЛУїШЗЃЌПЩФмОпгаПЙЖЏТіжрбљгВЛЏЕФзїгУЁЃЦфЫћGLP-1RAsЖдаФбЊЙмМВВЁЕФБЃЛЄзїгУВЛУїЯдЃЌЕЋЖдЦфЫћАВШЋВЮЪ§УЛгагаКІгАЯьЃЌвђДЫGLP-1RAsЕФЗчЯе-аЇвцЗжВМЪЧОљКтЕФЁЃGLP-1RAs Жд 2 аЭЬЧФђВЁЛМепЕФЗЪХжжЂзДвВБэЯжГіжЮСЦзїгУЁЃThondam БЈЕРЃЌвЛУћЛМгабЯжиЯТЧ№ФдЗЪХжКЭИїжжЗЪХжЯрЙиВЂЗЂжЂЃЈАќРЈ 2 аЭЬЧФђВЁЃЉЕФЛМепЖдАЌШћФЧыФЗДгІСМКУЃЌЬхжиКЭбЊЬЧПижЦУїЯдИФЩЦЃЌПЩФмЪЧЭЈЙ§діМгБЅИЙИаКЭМѕЩйФмСПЩуШыЕФжаЪрЕїНкЛњжЦЪЕЯжЕФЁЃвЛЯюЖд 25 УћЗЪХж 2 аЭЬЧФђВЁЛМепЕФбаОПБэУїЃЌНгЪмЖўМзЫЋывКЭЛЧѕЃыхРр/DPP-4 вжжЦМСжЮСЦ 6 ИідТЕФЛМепЃЌШчЙћЭЌЪБЗўгУ GLP-1RAЃЈАЌШћФЧыФ 19ЃЌ6 Р§ЃЉЃЌЦНОљЬхжиЁЂЬЧЛЏбЊКьЕААзКЭИЮФкжЌжЪОљЯджјНЕЕЭ. 25 УћНгЪмАЌШћФЧыФКЭРћРТГыФжЮСЦ 3 ИідТЕФ 25 Ућ 2 аЭЬЧФђВЁЛМепЕФЬхжижИЪ§КЭжЌЗОКёЖШвВЯджјЯТНЕ. 2 аЭЬЧФђВЁПЩЕМжТЙЧжЪДрЛЏЃЌдіМгЙЧелКЭЙЧелгњКЯВЛСМЕШЙЧЯрЙиВЂЗЂжЂЕФЗчЯеЁЃЪЕбщбаОПЗЂЯжЃЌGLP-1RAs ЖдЙЧжЪКЭЧПЖШгаЯджјЕФЛ§МЋзїгУЃЌПЩФмЪЧЭЈЙ§ИФЩЦЙЧїРНЁПЕЫљБиашЕФЙЧїРбЊвКЙЉгІЁЃвЛЯюбаОПНЋРћРТГыФгІгУгкТбГВЧаГ§ЕФ 2 аЭЬЧФђВЁДѓЪѓЃЌЫцКѓЖдЙЧЫшРДдДЕФЭтУкЬхЮЂаЁ RNA (miRNA) НјааИпЭЈСПВтађЁЃНсЙћБэУїЃЌРћРТГыФПЩЕМжТАаЯђвШЕКЫиаХКХЭЈТЗЕФЭтУкЬх miRNA ЗЂЩњЯджјБфЛЏЃЌЖјЙЧЫшЭтУкЬхНщЕМЕФ Wnt/ІТ-catenin аХКХЭЈТЗЕФБфЛЏгыЙЧБЃЛЄзїгУгаЙиЁЃ
GLP-1RAжЮСЦзюГЃМћЕФИБзїгУЪЧЮИГІЕРЯрЙиВЛСМЗДгІЃЈШчЖёаФЁЂХЛЭТКЭИЙаКЃЉКЭзЂЩфВПЮЛЗДгІЃЌЖјГЄаЇGLP-1RAИБзїгУНЯЩйЃЌИјвЉЦЕТЪНЯЕЭЃЌвРДгадИќКУЁЃЖўМзЫЋывШдЪЧСйДВжЮСЦ2аЭЬЧФђВЁЕФвЛЯпвЉЮяЁЃИљОнХЗжоЬЧФђВЁбаОПаЛсКЭУРЙњЬЧФђВЁаЛсЕФНЈвщЃЌЖдгкЕЅгУЖўМзЫЋывбЊЬЧПижЦВЛМбЕФЛМепЃЌНЈвщGLP-1RAЁЂЛЧѕЃыхРрЁЂрчпђЭщЖўЭЊРрЁЂDPP-4вжжЦМСЁЂФЦ-ЦЯЬбЬЧаЭЌзЊдЫЕААз2вжжЦМСКЭвШЕКЫизїЮЊВЙГфвЉЮяЃЌЕЋЛљгкGLP-1RAsГ§СЫПижЦбЊЬЧжЎЭтЛЙгажюЖрЦфЫћвцДІЃЌАќРЈБЃЛЄЩідрЁЂНЕЕЭаФбЊЙмМВВЁЗчЯеЁЂПижЦЬхжиЁЂЮоЕЭбЊЬЧЗчЯеЁЂЖдЙЧїРжЂзДгавцЁЂИБзїгУЗЂЩњТЪЕЭЕШЃЌGLP-1RAsдкЮДРДЕФT2DMжЮСЦжаБиНЋЗЂЛгВЛПЩЬцДњЕФзїгУЁЃ
жЮСЦадыФдкаФбЊЙмМВВЁжЮСЦжаЕФгІгУ
дкЗЧДЋШОадМВВЁжаЃЌаФбЊЙмМВВЁЪЧФПЧАШЋЧђЗЖЮЇФкЕМжТЫРЭіКЭЗЂВЁТЪзюИпЕФМВВЁЁЃИпбЊбЙЪЧаФбЊЙмМВВЁЕФжївЊЮЃЯевђЫижЎвЛЃЌБЛШЯЮЊЪЧгЩгкЩіЫи-бЊЙмНєеХЫи-ШЉЙЬЭЊЯЕЭГЃЈRAASЃЉКЭНЛИаЩёОЯЕЭГЛюадЙ§ИпвдМАФЦфѓСєЫљжТЁЃRAASжаЕФбЊЙмНєеХЫизЊЛЛУИЃЈACEЃЉЕФЙІФмЪЧНЋбЊЙмНєеХЫиIСбНтЮЊбЊЙмНєеХЫиIIЃЌЪЙбЊЙмЪеЫѕЃЌМфНгЩ§ИпбЊбЙЃЌЖјACE2дђНЋбЊЙмНєеХЫиIIЫЎНтЮЊбЊЙмРЉеХМСбЊЙмНєеХЫиЃЈ1-7ЃЉЃЌМфНгНЕбЊбЙЁЃвђДЫЃЌеыЖдRAASЪЧПижЦаФбЊЙмМВВЁЕФРэЯыВпТдЁЃКЯГЩЕФбЊЙмНєеХЫиIIгк2017ФъЛёЕУFDAХњзМЃЌгУгкЭЈЙ§ОВТіЪфзЂдіМгАмбЊжЂЛђЦфЫћЗжВМЪНанПЫГЩШЫЛМепЕФбЊбЙЁЃMontoneЕШШЫДгаБЩњЫФУцЬхЮЂдхжаЗжРыЩИбЁГіЕФЫФжжыФЃЈWPRGYFLЁЂGPDRPKFLGPFЁЂWYGPDRPKFLКЭSDWDRFЃЉЯдЪОГіЖдACEЕФвжжЦЛюадЁЃLiaoЕШШЫЗЂЯжРДздЕАЧхЕФШ§ыФIRWЭЈЙ§ЩЯЕїACE2ЕФБэДяРДНЕЕЭздЗЂадИпбЊбЙДѓЪѓЕФбЊбЙЁЃетаЉбаОПБэУївдRAASЮЊАаЕуЕФЪГЮяРДдДыФдкжЮСЦаФбЊЙмМВВЁЗНУцОпгаЧБдкЕФгІгУМлжЕЁЃ
РћФЦыФ (NPs) ЃЌАќРЈаФЗПРћФЦыФ (ANP)ЁЂФдРћФЦыФ (BNP) КЭ C аЭРћФЦыФ (CNP)ЃЌЪЧаФдрКЭбЊЙмЮШЬЌЕФживЊЕїНкМСЃЈЭМ12ЃЉЁЃвђДЫЃЌеыЖд NPs ЪЧдЄЗРЛђжЮСЦаФбЊЙмМВВЁЕФСэвЛжжЪЕгУВпТдЁЃФЮЮїСЂыФЪЧвЛжжжизщШЫ BNPЃЌ2001 ФъБЛ FDA ХњзМгУгкжЮСЦОВЯЂЛђЧсЖШКєЮќРЇФбЛМепЕФМБадЪЇДњГЅадаФСІЫЅНпЃЛШЛЖјЃЌгЩгкЦфЬивьадЕЭКЭАВШЋадгаЯоЃЌЫќЩаЮДЕУЕНЙуЗКгІгУЁЃNPs жївЊЭЈЙ§ NPR-A КЭ/Лђ NPR-B ЪмЬхЦ№зїгУЃЌЖј NPR-C жївЊгУгкЧхГ§ NPsЁЃCenderitide ЪЧвЛжжЫЋжи NPR-A/NPR-B МЄЖЏМСЃЌгЩ CNP КЭДгТЬТќАЭЩпжаЗжРыЕФЪїблОЕЩпРћФЦыФЕФ C ЖЫзщГЩЁЃCenderitide ФПЧАе§ДІгкСйДВбаОПжаЃЌВЂвбЯдЪОГіжЮСЦаФСІЫЅНпКЭЩіЫЅНпЕФАВШЋадКЭЧБСІЁЃДЫЭтЃЌвЛаЉЖдаФбЊЙмМВВЁгавцЕФыФе§дкЖЏЮяЩэЩЯНјааВтЪдЁЃР§ШчЃЌдкДѓЪѓжаЪфзЂбЊЙмЛюадГІыФПЩдіМгаФМЁбЊЙмЛюадГІыФЕФХЈЖШВЂФцзЊЯжгаЕФаФМЁЯЫЮЌЛЏЃЌЛЗыФ RD808 ПЩжаКЭ ІТ 1 -ЩіЩЯЯйЫиФмЪмЬхЃЌДгЖјМѕЧс ІТ 1-ЩіЩЯЯйЫиЪмЬхЁЃжаЪрДйЩіЩЯЯйЦЄжЪМЄЫиЪЭЗХвђзг (CRF) ЯрЙиыФЯЕЭГФПЧАзїЮЊдЄЗРаФбЊЙмМВВЁЕФАаЕуЪмЕНдНРДдНЖрЕФЙизЂЁЃCRFЯрЙиыФЯЕЭГгыаФбЊЙмЯЕЭГжЎМфДцдкИДдгЕФЙиЯЕЃЌЕЋЦфдкаФбЊЙмЙІФмжаЕФШЗЧаЕїНкзїгУШдгаД§ШЗЖЈЁЃДЫЭтЃЌдк 2 аЭЬЧФђВЁЛМепжаЃЌбЛЗ DPP-4 ЛюаддіМгЃЌбЊСїНщЕМРЉеХНЕЕЭЁЃбЊСїНщЕМРЉеХЪЧЙЋШЯЕФФкЦЄЙІФмеЯАЕФЬцДњБъжОЮяЃЌвВЪЧЮДРДаФбЊЙмЪТМўЕФдЄВтвђзгЃЌетБэУї DPP-4 ПЩФмЪЧдЄЗРаФбЊЙмМВВЁЕФЧБдкАаЕуЁЃ
жЮСЦадыФдкЮИГІЕРМВВЁжЮСЦжаЕФгІгУ
жЮСЦадыФдкГІЕРМВВЁжЮСЦжаЕФгІгУ
дкШЫЬхжаЃЌЮИГІЕРОњШКЙЙГЩИДдгЕФЮЂЩњЬЌЯЕЭГЁЃЭЈГЃЃЌШЫЬхЮИГІЕРОњШКЙЙГЩИДдгЕФЮЂЩњЬЌЯЕЭГЁЃЭЈГЃЃЌЩЯЦЄЭЈЙ§ЬсЙЉЮяРэЦСеЯКЭЗжУкИїжжПЙОњвђзгЃЌАќРЈПЙОњыФЃЈAMPsЃЉРДЕїНкГІ№ЄФЄНчУцГІЕРОњШКЕФзщГЩЁЃгХЪЦОњШКЃЈЩњРэОњШКЃЉКЭШѕОњШКЃЈжТВЁОњЃЉБЃГжЖЏЬЌЦНКтЃЌдкЭтРДЯИОњЁЂВЁЖОЁЂМФЩњГцЁЂЪГЮяжаЖОЁЂвЉЮяВЛСМЗДгІЁЂвХДЋвђЫиЕШв§Ц№ЕФИїжжГІЕРМВВЁжаЃЌШчГІбзЁЂБуУиЁЂГІРЃбёЁЂбзжЂадГІВЁЃЈIBDЃЉЕШЃЌЙВЩњОњЕФЖрбљадОљБЛЦЦЛЕЁЃПЙЩњЫиЕФДѓСПЪЙгУПЩФмНјвЛВННЕЕЭЙВЩњОњЕФЩњЮяЖрбљадЃЌВЛРћгкжЮСЦЃЌЩѕжСПЩФмМгжиВЁЧщЃЌР§ШчIBDЛМепдкШЗеяЧА2-5ФъФкИќгаПЩФмЪЙгУЙ§ПЙЩњЫиЁЃЖрыФвЉЮявђЬивьадЁЂгааЇадЁЂЕЭЖОадЕШЬиЕуЃЌдкИУСьгђв§Ц№СЫЙуЗКЙизЂЁЃ
ГІЕРе§ГЃОњШКЕФЯджјИФБфКЭЫожї-ЮЂЩњЮяЙВЩњЙиЯЕЕФЦЦЛЕПЩФмЪЧ IBD ЗЂЩњЗЂеЙЕФЙиМќЁЃIBD АќРЈПЫТоЖїВЁКЭРЃбёадНсГІбзЃЌЪЧгЩГІЕРУтвпЗДгІв§Ц№ЕФЃЌЯрЙибзжЂЪЧгЩЛЗОГКЭвХДЋвђЫиЯрЛЅзїгУв§Ц№ЕФЁЃЕЋ IBD ЕФОпЬхЗЂВЁЛњжЦЩаВЛУїШЗЃЌФПЧАЩаЮогааЇЕФжЮСЦЗНЗЈЁЃIBD ЛМепГІЕРЮЂЩњЮяЖрбљадУїЯдНЕЕЭЃЌСНДѓгХЪЦУХКёБкОњУХЃЈУЋТнОњПЦЃЉКЭФтИЫОњУХУїЯдМѕЩйЃЌЖјБфаЮИЫОњУХУїЯддіЖр. ДѓСПжЄОнБэУїБфаЮИЫОњУХГЩдБдк IBDжаЗЂЛгЙиМќзїгУЁЃИЌАБЫс-ОЋАБЫс-39 ЪЧвЛжжгЩжэЙЧЫшКЭСмАЭзщжЏздШЛЗжУкЕФаЁбєРызг AMPЃЌвбжЄЪЕОпгаПЙОњЁЂУтвпЕїНкКЭГІЩЯЦЄаоИДЙІФмЃЌПЩФмЮЊ IBDЬсЙЉАВШЋЕФЬцДњСЦЗЈЁЃ
ПЫТоЖїВЁЛМепЭЈГЃНгЪмГІЧаГ§ЪѕжЮСЦЃЌЕМжТЖЬГІзлКЯеїЃЈSBSЃЉЁЃаЁГІЫ№ЩЫКЭГіЩњЪБаЁГІвьГЃЖЬаЁвВПЩФмЕМжТ SBSЃЌЦфЖЈвхЮЊгыВаСєаЁГІГЄЖШГжајаЁгк 200 cm ЯрЙиЕФжЂзДЁЃGLP-2 гЩГІФкЗжУк L ЯИАћКЭжаЪрЩёОЯЕЭГЕФИїжжЩёОдЊВњЩњЃЈЭМ13ЃЉЃЌНќФъРДЃЌGLP-2дкSBSжЮСЦжаЪмЕНЙуЗКЙизЂЁЃGLP-2вбБЛжЄЪЕОпгаЖржжгавцзїгУЃЌАќРЈДЬМЄГІвўЮбЯИАћЩњГЄЁЂМѕЩйГІЯИАћЕђЭіЁЂДйНјГІ№ЄФЄРЉеХЁЂвжжЦЮИЫсЗжУкКЭЮИХХПеЁЂДЬМЄГІЕРбЊСїЁЂдіЧПГІЕРЦСеЯЙІФмЁЂМѕЧсбзжЂЫ№ЩЫЁЂДйНјгЊбјКЭвКЬхЮќЪеЁЃGLP - 2ЛЙЕїНкАБЛљЫсзЊдЫЕААзЕФБэДяЃЌжБНгМЄЛюmTORC1ЃЌдіМгГІЕРЩЯЦЄЖдАБЛљЫсЕФЮќЪеЁЃвЛаЉЬиЖЈЕФАБЛљЫсЃЈАќРЈЙШАБѕЃАЗЁЂЙШАБЫсЁЂОЋАБЫсЁЂИЪАБЫсЁЂРЕАБЫсЁЂЕААБЫсКЭКЌСђАБЛљЫсЃЉвВБЛжЄУїдкЮЌГжГІЕРЭъећадЗНУцЗЂЛгживЊзїгУЃЌАќРЈЗРжЙГІЕРЮЎЫѕЁЂИФЩЦГІЕРЦСеЯЙІФмЁЂМѕЩйбзжЂКЭЯИАћЕђЭіЁЃФкдДадGLP-2взБЛDPP-4НЕНтЃЌЕЋGLP-2РрЫЦЮяЬцЖШТГыФЭЈЙ§НЋGLP-2 NЖЫЕкЖўЮЛЕФБћАБЫсЬцЛЛЮЊИЪАБЫсЃЌНЋАыЫЅЦкДг7ЗжжгбгГЄжСдМ2-3аЁЪБЃЌгааЇзшжЙЦфБЛDPP-4НЕНтЁЃСйДВбаОПБэУїЃЌЬцЖШТГыФПЩгааЇМѕЩйЛђЯћГ§ЖдГІЭтгЊбјКЭ/ЛђОВТіЪфвКжЇГжЕФашвЊЃЌЖјЬцЖШТГыФдкдЖЖЫЛиГІЧаГ§ЕФгзжэжагІгУдђЯджјдіМгВаГІЕЅЮЛжиСПКЭЕААзжЪКЯГЩЁЃЬцЖШТГыФгк2012ФъБЛFDAХњзМгУгкSBSЛМепЕФСйДВжЮСЦЁЃWiśniewskiЕШШЫЩшМЦСЫвЛЯЕСаGLP-2РрЫЦЮяЃЌАќРЈ2-ИЪАБЫсШЁДњЁЂ10-е§ССАБЫсШЁДњЁЂ11-КЭ/Лђ16-ЪшЫЎШЁДњЕШЃЌЦфжааэЖрЖдGLP-2RЕФСЦаЇгХгкЬьШЛМЄЫиЃЌБэЯжГіСМКУЕФЪмЬхбЁдёадКЭЕЭЯЕЭГЧхГ§ТЪЁЃЦфжаЃЌыФЖЮЃЈ([2Gly, 10Nle, 11DPhe, 16Leu] hGLP-2-(1−33)-NH2 ЃЉБЛбЁЮЊСйДВПЊЗЂКђбЁвЉЮя. вШИпбЊЬЧЫидМвзхжаЕФ GLP-1 Опгагы GLP-2 ЯрЫЦЕФЙІФмЃЌВЂвбБЛНЈвщгУгкжЮСЦ SBSЁЃдквЛЯюбаОПжаЃЌЮхУћ SBS ЛМепдкНгЪм GLP-1 МЄЖЏМСАЌШћФЧыФжЮСЦКѓЃЌХХБуЦЕТЪКЭаЮЬЌОљгаЫљИФЩЦЁЃЭЌбљЃЌGLP-1 МѕЧсСЫОХУћ SBS ЛМепЕФИЙаКЃЌЕЋаЇЙћВЛШч GLP-2ЃЌЖј GLP-1 КЭ GLP-2 ЕФзщКЯгХгкЕЅЖРЪЙгУЦфжаШЮКЮвЛжжЁЃвШИпбЊЬЧЫидМвзхЕФСэвЛИіГЩдБГІДйвШЕКЫиЫЦКѕвВВЮгыСЫаэЖрЙ§ГЬЃЌР§ШчГІФкгЊбјЁЂдЫЖЏКЭЮИЫсЗжУкЃЌетБэУїПЊЗЂГІДйвШЕКЫибљыФЕФЧАОА. ЦфЫћЩњГЄвђзгШч EGFЁЂДйКьЯИАћЩњГЩЫиКЭИЮЯИАћЩњГЄвђзгвВвбЯдЪОГіЖд SBS ЕФжЮСЦЧБСІЁЃEGF КЭ GLP-2 ЕФзщКЯдіМгСЫШ§жж SBS аТЩњжэФЃаЭЕФаЁГІГЄЖШЃЌБэУї EGF ОпгажЮСЦаТЩњЖљ SBS ЕФЧБСІЁЃДйКьЯИАћЩњГЩЫиЭЈЙ§ДЬМЄЖЏЮяФЃаЭжаНєУмСЌНгЕААзЕФБэДяРДБЃЛЄГІЕРЦСеЯЙІФмВЂБЃЛЄЮИГІЕРУтЪмШБбЊ/дйЙрзЂЫ№ЩЫЃЌГІФкзЂЩфИЮЯИАћЩњГЄвђзгПЩНЕЕЭДѓЪѓЛЕЫРадаЁГІНсГІбзЕФЗЂВЁТЪКЭбЯжиГЬЖШЁЃ
МшФбЫѓОњжТВЁОњжъВњЩњЕФМшФбЫѓОњЖОЫиAПЩв§Ц№ИаШОепЕФИЙаККЭбзжЂЃЌЩѕжСбЯжиЕФЮБФЄадНсГІбзЁЃДѓѓЙЫи-2ЃЈYPCKLNLKLGKVPFHЃЉЪЧJiЕШДгУРжоДѓѓЙжаЗжРыЕУЕНЕФAMPЃЌПЩзшЖЯМшФбЫѓОњЖОЫиAв§Ц№ЕФ№ЄФЄЫ№ЩЫКЭбзжЂ ЃЌЪЧНќЦкБЛШЯЖЈЮЊЛКНт/жЮСЦМшФбЫѓОњЖОЫиAв§Ц№ЕФЮБФЄадНсГІбзЕФКђбЁвЉЮя. ДгКЋЙњђођыжаЗжРыГіЕФ 9 ОлЬхЖўСђМќЖўОлЬхыФ CopA3 (LLCIALRKK) ЯджјИФЩЦСЫМшФбЫѓОњЖОЫиA в§Ц№ЕФаЁГІбзжЂЗДгІЃЈМБадГІбзЃЉЃЌВЂЭъШЋзшЖЯСЫаЁЪѓЦЯОлЬЧСђЫсФЦв§Ц№ЕФТ§адНсГІбзЕФбзжЂЗДгІКЭЫцКѓЕФжТУќЗДгІ ЁЃВњЦјМдФЄЫѓОњA аЭв§Ц№ЕФЪГЮяжаЖОгыМИжжживЊЕФШЫРрЮИГІЕРМВВЁгаЙиЃЌБЛШЯЮЊЪЧгЩВњЦјМдФЄЫѓОњЕФВњЩњНщЕМЕФГІЖОЫи (CPE) гыШЫГІЕРНєУмСЌНгЕААзНсКЯЁЃArchana ЕШШЫЗЂЯжЃЌНЋ CPE гыНєУмСЌНгЕААз-4 АћЭтЛЗ ECL-2 ыФдЄЗѕг§ЛђЙВЗѕг§ПЩЯджјвжжЦ CPE геЕМЕФЭУГІЛЗ344ЧЛФквКЬхЛ§ОлКЭзщжЏбЇЫ№ЩЫЃЌетБэУїКЯГЩыФ ECL-2 ПЩФмДњБэвЛжждЄЗРA аЭВњЦјМдФЄЫѓОњв§Ц№ЕФГІЕРзщжЏбЇЫ№ЩЫЕФЧБдкВпТдЁЃШЫНсГІЩЯЦЄЗжУкЕФПЙОњыФЪЧСэвЛжжОпгаЙуЗКПЙОњКЭУтвпЕїНкЙІФмЕФ AMPЁЃзюНќЕФбаОПБэУїЃЌШЫПЙОњыФгажњгкдчЦкНсГІЩЯЦЄЯИАћЕжгљГІдДадЪѓЩЫКЎЩГУХЪЯОњзшжЙЯИОњШыЧжЁЂЮЌГжЩЯЦЄЦСеЯЕФЭъећадЃЌПЩФмЭЈЙ§ВњЩњTollбљЪмЬх4КЭДйбзЯИАћвђзгРДЪЕЯжЁЃДЫЭтЃЌГІЕРВЁЖОИаШОвВБЛжЄУїФмДЬМЄAMPsЕФБэДяЁЃГТЕШбаОПЗЂЯжаЁКЫЬЧКЫЫсВЁЖОИаШОдіМгГІЩЯЦЄЯИАћШЫІТЗРгљЫи3ЕФБэДяКЭЗжУкЃЌШЫІТЗРгљЫи3ОпгаЯИАћЭтПЙГІЕРВЁЖОЛюадЁЃ
ФвадЯЫЮЌЛЏ (CF) ЛМепЭЈГЃЛЙЛсГіЯжГІЙЃзшКЭБуУиЃЌПЩФмЗЂеЙЮЊдЖЖЫГІЙЃзшзлКЯеїЁЃФёмеЫсЛЗЛЏУИ C (GCC) ЪмЬхМЄЖЏМСРћФЧТхыФгк 2012 ФъЛё FDA ХњзМгУгкжЮСЦТ§адБуУиЁЃРћФЧТхыФЛЙБЛжЄУїПЩвдИФЩЦ CF ФЃаЭаЁЪѓЕФГІЕРдЫЪфЃЌОЁЙмЛЙашвЊНјвЛВНбаОПРДЦРЙРЦфЖд CF ЛМепГІЕРВЁРэЕФгАЯьЁЃNHE3 вжжЦМС tenapanor ЭЈЙ§вжжЦФЦЮќЪеРДИФЩЦ CF аЁЪѓЕФЮИГІЕРдЫЪфЃЌетБэУївжжЦ GCC аХКХзЊЕМКЭ NHE3ПЩФмЪЧжЮСЦ CF ЛМепБуУиЕФКЯЪЪАаЕуЁЃ
Г§СЫвЉЮябмЩњыФЭтЃЌыФвВПЩФмРДздЪГЮяЁЃAsn-Pro-Trp-Asp-Gln (NPWDQ) ЪЧвЛжжЭЈЙ§ЫЎНтРвЕААзЃЈвЛжжжївЊЕФХЃФЬЕААзЃЉЛёЕУЕФыФЃЌЫќЯджјвжжЦСЫЪГЮяЙ§УєдТбЧхЕААзЩјЭИЕНШЫЬхГІЕР Caco-2 ЯИАћжаЃЌетБэУїетжжыФПЩФмИФЩЦГІЕРЩЯЦЄЦСеЯЕФЙІФмЁЃІТ-Casofensin ЪЧвЛжждкЗЂНЭШщжаЗЂЯжЕФыФЃЌЬхФкЪЕбщЗЂЯжЃЌдчЦкЗўгУ ІТ-Casofensin ПЩЭЈЙ§БЃЛЄБзДЯИАћКЭДйНјЩЫПкгњКЯРДМѕЩйпХпсУРаСв§Ц№ЕФГІЕРЫ№ЩЫКЭбзжЂЁЃпХпсУРаСв§Ц№ЕФГІЕРЫ№ЩЫОпгагыПЫТоЖїВЁЯрЭЌЕФСйДВЁЂзщжЏбЇКЭВЁРэЩњРэбЇЬиеїЬсЪОЃЌІТ-casofensinПЩФмЪЧПЫТоЖїВЁЕФЧБдкИЈжњжЮСЦЗНЗЈЁЃ
ЖрыФвЉЮядкаѓЧнГІЕРМВВЁжЮСЦжавВгазХЙуРЋЕФЧАОАЃЌСѕЕШвдЬьШЛЩпЕЈМюЮЊдСЯЃЌбажЦГіИФСМКЯГЩыФKR-32ЃЌKR-32ПЩИФЩЦВњГІЖОЫиДѓГІИЫОњK88ИаШОзажэЕФжЌЗОЫсЮќЪеВЛСМЁЂввУбЬсШЁЮязмЯћЛЏТЪМАГІЕРаЮЬЌЃЌЬсЪОKR-32ОпгаЧБдкЕФвЉгУМлжЕЁЃC-BFЪЧвЛжжРДдДгкПЙОњыФЕФЖрыФЃЌЪЧAMPМвзхжазюЮЊЭЛГіЕФвЛИіЃЌБЛШЯЮЊЪЧзюгаЧАЭОЕФПЙЩњЫиЬцДњЦЗЁЃC-BFЯджјИФЩЦСЫЖЯФЬзажэЕФЩњГЄЃЌИФЩЦСЫLPSЖдаЁГІЕФНсЙЙКЭЗЂг§Ы№ЩЫЃЌетБэУїC-BFПЩФмЪЧжЮСЦLPS/ВЁдЬхв§Ц№ЕФГІЕРЫ№ЩЫЕФЧБдквЉЮяЁЃ
жЮСЦадыФдкЮИВЁжЮСЦжаЕФгІгУ
ЮИ№ЄФЄЪЧШЫРрКЭЖЏЮязюДрШѕЕФзщжЏжЎвЛЃЌЮИВЁЪЧГЃМћЕФЮЪЬтЁЃгФУХТнИЫОњИаШОЁЂЗЧчоЬхПЙбзвЉЁЂОЦОЋЁЂЮќбЬЁЂЧщаїКЭбЙСІЪЧдьГЩЮИЫ№ЩЫЕФжївЊвђЫиЃЌНјЖјЕМжТЮИбзКЭРЃбёЁЃЮИВЁШєВЛМАЪБжЮСЦЛђжЮСЦВЛЕБЃЌЛсЗЂеЙГЩТ§адВЁЃЌГжајЕФГЄЦкЫ№ЩЫЛсДѓДѓдіМгЮИАЉЕФЗчЯеЁЃЮИАЉЪЧФПЧАШЋЧђЕкЫФДѓГЃМћАЉжЂЃЌдкФаадКЭХЎадАЉжЂЫРЭідвђжаЗжБ№ХХУћЕкШ§КЭЕкЮхЁЃ
ЫфШЛФПЧАЛЙУЛгаыФРрвЉЮяБЛХњзМгУгкжЮСЦЮИВЁЃЌЕЋНќЪЎФъРДЃЌыФРрвЉЮяЃЌАќРЈФкдДадКЭЭтдДадыФРрвЉЮядкЮИВЁжаЕФзїгУЕУЕНСЫЙуЗКЕФбаОПЁЃНЕИЦЫиЛљвђЯрЙиыФЃЈCGRPЃЉЙуЗКЗжВМгкЮИГІЕРЯЕЭГЃЌЪЧРБНЗЫиУєИаИаОѕЩёОЕФжївЊЩёОЕнжЪЁЃетаЉИаОѕЩёОВЮгыБЃЛЄЮИеГФЄУтЪмИїжжДЬМЄЃЌCGRPдкДЫЙ§ГЬжаЦ№ЧБдкНщжЪзїгУЃЌдіМгЮИеГФЄбЊСїЃЌвжжЦЮИЫсЗжУкЃЌЗРжЙЯИАћЕђЭіКЭбѕЛЏЫ№ЩЫЁЃГ§CGRPЭтЃЌвЛбѕЛЏЕЊКЯУИ-вЛбѕЛЏЕЊЃЈNOS-NOЃЉКЭЛЗбѕКЯУИ-ЧАСаЯйЫиЃЈCOX-PGЃЉЯЕЭГЖдЮИвВгаРрЫЦЕФБЃЛЄзїгУЁЃCGRPЁЂNO КЭ PG БЛШЯЮЊЪЧЮИБЃЛЄЕФжеЖЫНщжЪЃЌВЂНщЕМаэЖрФкдДадыФЕФЮИБЃЛЄзїгУЁЃввДМв§Ц№ЕФЮИЫ№ЩЫЕФжївЊЗЂВЁЛњжЦЪЧЮИЮЂбЊЙмЫ№ЩЫЁЃзїЮЊЩёОЩњГЄвђзггеЕМ (VGF) ЛљвђбмЩњЕФыФЃЌTLQP-21 гЩзщГЩад NOЁЂPGE2 КЭЩњГЄвжЫиНщЕМЃЌБэУїжаЪрЖјЗЧЭтжмзЂЩфПЩвдвдМССПвРРЕадЗНЪНМѕЧсввДМв§Ц№ЕФЮИЫ№ЩЫ, Novokinin (Arg-Pro-Leu-Lys-Pro-Trp) ЪЧгЩТбМЄыФаоЪЮЕФгааЇбЊЙмРЉеХКЭПЙИпбЊбЙыФЃЌЖдбЊЙмНєеХЫи II 2 аЭ (AT2) ЪмЬхгаНЯИпЕФбЁдёадЧзКЭСІЁЃеХЕШбаОПЗЂЯжЃЌnovokinin вдМССПвРРЕадЗНЪНвжжЦФдЪвФкзЂЩфКѓЛљДЁЮИЫсЗжУкЃЌБЃЛЄЮИ№ЄФЄУтЪмОЦОЋв§Ц№ЕФЫ№ЩЫЃЌетЪЧЭЈЙ§НщЕМ AT2 ЪмЬх-PG ЭЈТЗЁЃетаЉНсЙћЬсЪОСЫ TLQP-21 КЭ novokinin дкЮИЫ№ЩЫжЮСЦжаЕФМлжЕЁЃДгЗЯЦЁОЦНЭФИЕААзЫЎНтЮя (гШЦфЪЧ < 3 kDa) жаЛёЕУЕФЖрыФЬсШЁЮяПЩМѕЧсДѓЪѓЮИ№ЄФЄЫ№ЩЫЃЌЬсЪОНЭФИЖрыФЬсШЁЮядкЮИВЁжЮСЦжаЕФЧБдкМлжЕЁЃ
ЖЏЮягІМЄадЮИЫ№ЩЫГЃБЛгУзїбаОПгІМЄадЮИВЁЛњжЦЕФФЃаЭЃЌAMPЬњЕїЫиБЛШЯЮЊЪЧгЩБкЯИАћЕїНкЮИЫсЩњГЩЖјВњЩњЕФЃЌЧУГ§ЬњЕїЫиЕФаЁЪѓЮИЫсЗжУкУїЯдМѕЩйЃЌЬсЪОЬњЕїЫиПЩФмгыгІМЄЬѕМўЯТЮИРЃбёЕФЗЂЩњгаЙиЁЃnesfatin-1ЪєгкбсЪГыФМвзхЃЌДцдкгкГІЕРЕФЩёОдЊКЭФкЗжУкЯИАћжаЁЃAlexandraЕШЕФбаОПБэУїЃЌnesfatin-1ЖдЫЎНўЪјИПгІМЄДѓЪѓЮИгаУїЯдЕФБЃЛЄзїгУЃЌЦфЛњжЦгыЮИвКЗжУкМѕЩйЁЂCOX-PGКЭNOS-NOЯЕЭГНщЕМЕФГфбЊЁЂУдзпЩёОЁЂИаОѕЩёОМАЯуВнШЉЪмЬхЕФМЄЛюгаЙиЁЃТ§адЮТКЭгІМЄПЩв§Ц№ДѓЪѓЮИРЃбёЃЌЩњГЄвжЫиРрЫЦЮяАТЧњыФПЩЭЈЙ§вжжЦЯИАћЕђЭіЁЂбзжЂКЭбѕЛЏРДМѕЧсЮИРЃ.жаЪрЖјЗЧЭтжмзЂЩфДпВњЫиФмЯћГ§ДѓЪѓЪјИПгІМЄв§Ц№ЕФВЭКѓЮИЪеЫѕдіЧПЃЌДгЖјМѕЩйЮИХХПебгГйЃЌЬсЪОДпВњЫиПЩФмЪЧжЮСЦгІМЄЯрЙиЮИГІЖЏСІеЯАЕФКђбЁвЉЮяЁЃ
ЮИАЉЪЧвЛжжбЯжиЕФЮИВПМВВЁЃЌЖржжЖрыФдкЮИАЉжЮСЦжаЯдЪОГіСМКУЕФЧАОАЁЃGEBP11ЪЧЭЈЙ§ЪЩОњЬхеЙЪОММЪѕЩИбЁКЭМјЖЈЕФвЛжжаТЕФОХАБЛљЫсЙщГВыФЁЃGEBP11бЁдёадЕигыШЫЦъОВТіФкЦЄЯИАћКЭжзСібЊЙмНсКЯЃЌЬсЪОЦфПЩФмЪЧжзСіЯдЯёКЭАаЯђвЉЮяЕнЫЭЕФживЊКђбЁвЉЮяЁЃЕт131БъМЧЕФЫЋЦчPEGЛЏGEBP11Ш§ОлЬхЃЈ131Imur2PEG- (GEBP11) 3ЃЉжЮСЦПЩЯджјвжжЦТуЪѓШЫЮИАЉвьжжвЦжВСіЕФЩњГЄЃЌбгГЄЩњДцЪБМфЃЌЬсЪО131Imur2PEG- (GEBP11) 3ПЩФмЪЧЮИАЉыФАаЯђжЮСЦЕФКЯЪЪКђбЁЮяЃЌвВЪЧЮИАЉПЙбЊЙмЩњГЩжЮСЦЕФвЉЮядиЬхЁЃгФУХТнИЫОњИаШОЪЧЮИАЉЕФживЊВЁвђжЎвЛЁЃHP-6ЃЈPro-Gln-Pro-Lys-Val-Leu-Asp-SerЃЉЪЧДгвТдхЮЂЩњЮяЫЎНтВњЮяжаЗжРыЕФЛюадыФЃЌвбБЛжЄУїОпгаЕжПЙгФУХТнИЫОњгеЕМЕФжТАЉадЁЃHP-6ЭЈЙ§вжжЦEGFRЛюЛЏЃЌЯТЕїСзЫсМЁДМ3-МЄУИ/AktаХКХзЊЕМКЭІТ-cateninКЫзЊЮЛЃЌгааЇвжжЦгФУХТнИЫОњгеЕМЕФШЫЮИЯйАЉЯИАћЃЈAGSЃЉдіжГКЭЧЈвЦЃЌЖјВЛвжжЦЯИОњЛюСІЛђAGSЯИАћЧжЯЎЁЃZhangЕШШЫКЯГЩСЫAMP pexigananМАЦфФЩУзПХСЃЃЈPNPsЃЉЃЌЦфБэЯжГіПЙгФУХТнИЫОњЛюадЃЌВЂЧвБШpexigananЖдаЁЪѓЮИжаЕФгФУХТнИЫОњгаИќЧПЕФЧхГ§ФмСІЃЌВЂЯдЪОГіжЮСЦКЭдЄЗРгФУХТнИЫОњЯрЙиЮИВЁЕФЧБСІЁЃTFF1ЪЧвЛжжгы№ЄЕААзЯрЙиЕФЮИ№ЄФЄЯИАћЗжУкыФЁЃTFF1ЕФБэДядкгФУХТнИЫОњИаШОЕФМБадЦкЖјЗЧТ§адЦкдкЮИёМжаЩЯЕїаЁЪѓИаШОЃЌВЂЧвгыбзжЂЗДгІГЪИКЯрЙиЃЌБэУї TFF1 ПЩФмгажњгкЯИАћЕжПЙЯИОњКЭТ§адбзжЂЕФЗЂеЙЁЃЭЌЪєвЛИіМвзхЕФ TFF2 вбБЛжЄУїФмгыЮИУкЫи MUC6 ЯрЛЅзїгУЃЌЮШЖЈЮИ№ЄвКЦСеЯЃЌЮЌГжЮИ№ЄФЄЭъећадЁЃ
ЖрыФРрвЉЮядкЮИЖЏСІЗНУцвВгаЕїНкзїгУЁЃЭтжмзЂЩфGLP-2ЭЈЙ§діМгCGRPКЭФкдДадPGsЖјЗЧNOРДдіМгЮИГІЕРбЊСїСПКЭЮИеГФЄбЊСїСПЁЃЭтдДадGLP-1дкРыЬхШЋЮИФЃаЭжаЭЈЙ§ЩёОв§Ц№NOЪЭЗХЕНЮИёМЃЌДгЖјНЕЕЭаЁЪѓЮИЖЏСІЁЃЕЋвбЛёХњЕФGLP-1/2бмЩњыФЪЧЗёОпгаРрЫЦзїгУШдгаД§баОПЁЃBNPОпгаРЉеХбЊЙмЕФЬиадЃЌПЩвддіМгФкдрЙрзЂКЭбѕКЯЃЌжизщBNPвбБЛжЄЪЕФмдіМгЮИеГФЄЮЂбЊЙмжаЕФбЊКьЕААзбѕКЯ. ЮИЖЏЫиКЭЩњГЄЫиЪЭЗХыФЪєгкЭЌвЛыФМвзхЃЌетаЉМЄЫидкЮИГІЕРдЫЖЏЕїНкжаЦ№зХживЊзїгУЁЃЩњГЄЫиЪЭЗХыФКЭЮИЖЏЫиПЩвддкЬхЭтКЭЬхФкаЭЌДЬМЄЧПСвЕФЮИЪеЫѕЁЃЮИЖЏЫивдМАЮИЖЏЫиКЭЩњГЄЫиЪЭЗХыФЕФНсКЯЭЈЙ§зщАЗНщЕМЕФЭООЖДЬМЄќќ ЮИЫсЗжУкЁЃ
жЮСЦадыФдкАЉжЂжЮСЦжаЕФгІгУ
ДЋЭГЕФАЉжЂжЮСЦЗНЗЈАќРЈЪжЪѕКЭЗХСЦЃЌЖдЭэЦкАЉжЂЛМепСЦаЇгаЯоЁЃЫцКѓАаЯђжЮСЦКЭУтвпжЮСЦЕФЗЂеЙЃЌДѓДѓЬсИпСЫАЉжЂЛМепЕФЩњДцТЪЁЃАаЯђжЮСЦРћгУжзСіЯИАћЖдЬиЖЈЗжзгЛђаХКХЭЈТЗЕФвРРЕЃЌвдЁАЕМЕЏЁБЕФЗНЪНЩБЫРжзСіЯИАћЁЃУтвпжЮСЦвЉЮяВЂВЛжБНгЙЅЛїжзСіЯИАћЃЌЖјЪЧЕїНкЛМепздЩэЕФУтвпЯЕЭГЃЌЭЈЙ§АаЯђУтвпМьВщЕуРДЙЅЛїжзСіЯИАћЃЌPD-1/PD-L1ЪЧжкЫљжмжЊЕФУтвпМьВщЕуЃЌФПЧАвбга5жжеыЖдPD-1/PD-L1ЯрЛЅзїгУЕФЕЅПЫТЁПЙЬхБЛFDAХњзМгУгкжЮСЦАЉжЂЃЌЕЋПЙЬхДцдкГЩБОИпЁЂПкЗўЪЪгІадВюЁЂУтвпдадЧПЕШШБЕуЁЃЖрыФгЩгкЗжзгГпДчаЁЁЂЧзКЭСІИпЁЂвзаоЪЮЁЂУтвпдадЕЭЕШгХЕуЃЌдкжзСіеяЖЯКЭжЮСЦСьгђвВв§Ц№СЫШЫУЧЕФЙизЂЃЌвЛаЉаоЪЮКѓЕФЖрыФЛЙБэЯжГіСМКУЕФЮШЖЈадЃЌР§ШчCarvajalЕШШЫПЊЗЂСЫЮШЖЈЕФІС-Тна§ыФзїЮЊMDM2КЭMDMXЕФвжжЦМСЃЌгУгкжЮСЦp53вРРЕадАЉжЂЁЃ
ЬьШЛыФдкЬхФкЕФАыЫЅЦкНЯЖЬЃЌеыЖджзСіЯИАћжаИїжжвьГЃБэДяЪмЬхЕФыФЭЈГЃЪЧОЙ§аоЪЮЕФыФРрЫЦЮяЁЃетаЉыФЕФЩњВњжївЊгаШ§жжЗНЗЈЃЌИїгагХШБЕуЃК1ЃЉДгЬьШЛЕААзжЪжабмЩњЃЛ2ЃЉЛЏбЇКЯГЩКЭЛљгкНсЙЙЕФКЯРэЙЄГЬЛЏЃЛ3ЃЉыФПтЩИбЁЁЃЦфжаЃЌЪЩОњЬхеЙЪОММЪѕЪЧвЛжжДЋЭГЧвгІгУЙуЗКЕФЗНЗЈЃЌОпгаВйзїМђЕЅЁЂФмгааЇЩИбЁДѓСПВЛЭЌыФЕФгХЕуЁЃ
ЖрыФдкжзСіжЮСЦжаЕФгІгУжївЊгаЫФжжЗНЪНЃК1ЃЉРћгУЗХЩфадЭЌЮЛЫиЁЂШОСЯЛђЦфЫћвбБЈЕРЕФЗжзгБъМЧЕФЖрыФзїЮЊЬНеыЃЌгУгкжзСіеяЖЯКЭГЩЯёЃЛ2ЃЉРћгУыФХМСЊЕФФЩУзВФСЯНјаажзСіжЮСЦЃЛ3ЃЉРћгУыФвпУчМЄЛюУтвпЯЕЭГНјаадЄЗРЃЛ4ЃЉЕЅЖРЪЙгУыФзїЮЊАаЯђвЉЮяЃЈЭМ14ЃЉЁЃ
ЛљгкыФЕФГЩЯёЬНеыгыжзСіжаЬивьадБэДяЕФЪмЬхНсКЯЁЃетаЉЪмЬхПЩвдБэДядкЯИАћБэУцЃЌР§Шч ІСvІТ3 ећКЯЫи (RGD ыФ)ЁЂEGF ЪмЬхЁЂЩњГЄвжЫиЪмЬхЁЂЩёОНЕбЙЫиЪмЬхКЭзЊЬњЕААзЪмЬхЃЛвВПЩвдБэДядкЯИАћФкЃЌР§Шч Bcr/AblЁЂЯИАћжмЦкЕААз A КЭЯИАћжмЦкЕААзМЄУИЃЛвВПЩвдБэДядкЯИАћЭтЛљжЪжаЃЌР§ШчЯЫСЌЕААзЁЂЛљжЪН№ЪєЕААзУИКЭЧАСаЯйЬивьадПЙдЁЃПЩвдЭЈЙ§ЕЅЙтзгЗЂЩфМЦЫуЛњЖЯВуЩЈУш/МЦЫуЛњЖЯВуЩЈУшПЩЪгЛЏЬНеыЕФЮЛжУвджИЪОжзСіЗжВМЁЃИУММЪѕвбгІгУгкдчЦкжзСіеяЖЯКЭЪжЪѕЧаГ§ЁЃвбОПЊЗЂСЫМИжжЬНеыЃЌР§ШчАТЧњыФКЭЕиЦеРћыФЃЌзїЮЊЩњГЄвжЫибљыФЕФЗХЩфадБъМЧНсКЯЮяЃЌвбБЛFDAХњзМгУгкжзСіГЩЯёЃЌР§ШчЩёОФкЗжУкжзСіКЭЗЮАЉ ЁЃВЛавЕФЪЧЃЌЕиЦеРћыФвбБЛГЗЛиЁЃЛљгкRGDыФЕФЗХЩфадБъМЧыФзюНќЪмЕНЙизЂЃЌВЂЧввбОКЯГЩСЫвЛЯЕСаЬНеыЃЌАќРЈЃЈ99mЃЉTc-3PRGD2ЃЌЫќПЩгУгкЭЈЙ§ЗХЩфадЕт387ЕФвѕадШЋЩэЩЈУшМьВтЗжЛЏаЭМззДЯйАЉ. ях 177 dotatate ЪЧвЛжжЗХЩфадБъМЧЕФЩњГЄвжЫиРрЫЦЮяЃЌзюНќБЛХњзМгУгкжЮСЦЩњГЄвжЫиЪмЬхбєадЕФЮИГІвШЩёОФкЗжУкжзСіЁЃЫќгыЩњГЄвжЫиЪмЬхНсКЯЃЌШЛКѓЪЭЗХЗХЩфадях 177 НјШыжзСіЯИАћЃЌЭЈЙ§аЮГЩЯИАћФкздгЩЛљгеЕМЯИАћЫ№ЩЫЁЃ
ЛљгкгыыФЛљЬНеыЯрЭЌЕФдРэЃЌПЩвдЭЈЙ§дкыФЩЯБъМЧІТЗЂЩфЬхРДЪЕЯжФкВПЗХЩфжЮСЦЃЛШЛЖјЃЌетУїЯдЪмЕНЖдОпгабєадЪмЬхЕФе§ГЃАазщжЏЕФЗјЩфЫ№ЩЫЕФЯожЦЃЌетаЉЪмЬхЮЛгкжзСіИННќЛђдЖДІЁЃвЛжжМѕЩйЧБдкИБзїгУЕФПЩФмЬцДњЗНЗЈЪЧНЋыФгыПЙАЉвЉЮяЁЂЛљвђКЭRNAЃЈаЁИЩШХRNAЃЈ[siRNA]/miRNA/mRNAЃЉЃЉХМСЊЕнЫЭЕНжзСіAN-152КЭAN-207ЪЧгыАЂУЙЫиХМСЊЕФДйЛЦЬхМЄЫиЪЭЗХМЄЫиРрЫЦЮяЃЌЖдДйЛЦЬхМЄЫиЪЭЗХМЄЫиЪмЬхбєадЕФАЉжЂОпгаПЙАЉЛюадЁЃIЦкКЭIIЦкСйДВбаОПНсЙћБэУїЃЌИУвЉЖдШщЯйАЉЁЂзгЙЌФкФЄАЉЁЂТбГВАЉЕФжЮСЦгааЇЃЌЖОадКЭИБзїгУжаЕШЁЃChen ЕШЩшМЦСЫОлввЖўДМЛЏжЌжЪЬх-ОлбєРызг-DNA (LPD) ФЩУзСЃЃЌЭЈЙ§аоЪЮАаЯђжзСіЬивьадЪмЬхАБЛљыФУИNЕФNGRыФЕУЕНLPD-PEG-NGRЁЃLPD-PEG-NGRвдШЋЩэадЁЂЬивьадЁЂИпаЇЕФЗНЪННЋsiRNAЕнЫЭжСаЁЪѓЪЕЬхСіФкЃЌВЂЧвЭЈЙ§ЕнЫЭc-myc siRNAЃЌЭЈЙ§ЯТЕїc-mycЕФБэДягааЇв§ЗЂжзСіЯИАћЕђЭіЃЌДгЖјвжжЦВПЗжжзСіЕФЩњГЄЁЃДЫЭтЃЌЭЈЙ§ЬхФкЪЩОњЬхЩИбЁЕУЕНЕФжзСіДЉЭИыФЃЌПЩвдгааЇНЋЙВМлХМСЊКЭЙВЭЌИјвЉЕФвЉЮяЕнЫЭжСжзСізщжЏЩюДІЁЃетаЉНсЙћБэУїЛљгкыФЕФвЉЮяЕнЫЭЯЕЭГЖджзСіЕФжЮСЦОпгаживЊЕФЧБСІЁЃ
РДздЬиЖЈАаЕААзЕФПЙдыФПЩвдзїЮЊПЙАЉыФвпУчЃЌЭЈЙ§гыПЙдГЪЕнЯИАћЩЯЕФжївЊзщжЏЯрШнадИДКЯЬх (MHC) НсКЯЃЌДЅЗЂИЈжњЛђЯИАћЖОад T ЯИАћЕФПЙжзСізїгУЁЃEGFRЃЌР§Шч EGFR1 КЭ HER2ЃЌЪЧжкЫљжмжЊЕФАЉжЂжЮСЦАаЕуЁЃЛљгк HER2 НсЙЙЕФыФвпУч TERT572Y БЛгУгк 46 УћЭэЦкЗЧаЁЯИАћЗЮАЉЛМепЁЃЦЄЯТзЂЩф TERT572Y ПЩгеЕМ TERT ЬивьадУтвпЗДгІВЂЯджјбгГЄЩњДцЦкЁЃManijehЕШЭЈЙ§PEPOPЗНЗЈМЦЫуВЂдЄВтЧБдкБэЮЛЃЌДгHER2АћЭтЧјжаЩИбЁГіЖржжыФађСазїЮЊКђбЁађСаЃЌЭЈЙ§ЗжзгЖдНгЦРЙРетаЉКђбЁыФгыMHC IРрКЭIIРрЗжзгЕФНсКЯЧзКЭСІЃЌбАевыФгыMHC IРрКЭIIРрЗжзгзюЮШЖЈЕФНсКЯНсЙЙЃЌЩИбЁГіMHC IРрКЭIIРрНсКЯыФзїЮЊШщЯйАЉыФвпУч396ЕЋДѓЖрЪ§ыФвпУчСйДВЪдбщЮДФмЯдЪОГігХвьЕФжЮСЦаЇЙћЃЌыФвпУчвВвђДЫКмЩйЪмЕНЙизЂЁЃВЛЙ§ЃЌTakumiЕШШЫШЯЮЊАќРЈыФдкФкЕФДѓЖрЪ§АЉжЂвпУчдкСйДВбаОПжаЮДФмГЩЙІЕФжївЊдвђЪЧЦфУтвпдадНЯВюЃЌВЂНЈвщгХЛЏыФЕФХфЗНЁЂзєМСКЭИјвЉЭООЖНЋЛёЕУРэЯыЕФаЇЙћЁЃыФБЛГЦЮЊЯИАћДЉЭИыФЃЈCPPЃЉЃЌвВПЩвдзїЮЊвЉЮядиЬхЃЌНЋЦфЫћыФЁЂЕААзжЪЁЂDNAЁЂаЁRNAКЭвЉЮядЫЫЭЕНЯИАћжаЁЃгЩNerinetideКЭCPP TatзщГЩЕФCPP-вЉЮяЙЙНЈЬхБЛгУгкНЋNerinetideЕнЫЭДЉЙ§бЊФдЦСеЯВЂНјШыЩёОдЊ. ыФЛЙЭЈЙ§гыПЙдНсКЯвдгеЕМПЙдЬивьадУтвпФЭЪмВЂНЕЕЭЭбАаЗДгІЕФЗчЯеЃЌЯдЪОГіСМКУЕФДЋЕнЙІФм . Tsoras ЪЙгУыФФЩУзДиРДИФЩЦыФбЧЕЅЮЛвпУчЖдАЉХпПЙдЕФУтвпдад .
ЖрыФГ§СЫзїЮЊвЉЮядиЬхЁЂвпУчЭтЃЌЛЙПЩвдЭЈЙ§гыАаЪмЬхНсКЯЗЂЛгПЙжзСізїгУЃЌЦфжазюЪмЙизЂЕФЪЧеыЖдPD-1/PD-L1аХКХЭЈТЗЕФЖрыФЁЃBoohakerЕШЩшМЦСЫвЛжжPD-L1ыФФЃФтЮяPL120131ЃЌИУыФПЩвдЭЈЙ§гыPD-1НсКЯРДИЩШХPD-1/PD-L1ЕФЯрЛЅзїгУЃЌдк3DЙВХрбјФЃаЭжаЃЌPL120131БШPD-1ПЙЬхИќФмЮЌГжЙВХрбјTЯИАћЕФДцЛюКЭЛюадЁЃZhouЕШЛљгкгыPD-1КЭPD-L1НсКЯЕФЖрыФЃЌЩшМЦСЫздвжжЦыФDS-IКЭDS-IIМАЦфЛЗыФаЮЪНЃЌЖдPD-1БэЯжГіКмЧПЕФЧзКЭСІЃЌAbbas ЕШШЫЩшМЦСЫвЛжжаТЕФАаЯђ PD-1 ЕФЖрыФFITC-YT-16ЃЌИУЖрыФдкЬхЭтЯджјдіЧПСЫTЯИАћЕФПЙжзСіЛюадЃЌЖј Sasikumar ЕШШЫЩшМЦСЫЖрыФ NP-12ЃЌгыPD-1ОКељадНсКЯ PD-L1ЁЃДЫЭтЃЌNP-12 дкКкЩЋЫиСіЁЂНсГІАЉКЭЩіЯИАћАЉЕФСйДВЧАФЃаЭжаЃЌдквжжЦдЗЂаджзСіЕФЩњГЄКЭзЊвЦЗНУцБэЯжГігыЩЬвЕЛЏ PD-1 АаЯђПЙЬхЯрЭЌЕФаЇЙћЁЃЫфШЛетаЉыФЛЙВЛЪЪКЯзшЖЯPD-1/PD-L1РДжЮСЦжзСіЃЌЕЋЫќУЧОпгаЙуРЋЕФЧАОАЁЃРДздЖЏЮяЕФЖОадыФЃЈVPЃЉвВПЩФмБэЯжГіПЙжзСізїгУЁЃгЩгкVPЬьШЛАаЯђВИШщЖЏЮяЪмЬхЃЌЫќУЧЖдЯИАћФЄЩЯЕФЬиЖЈРызгЭЈЕРКЭЪмЬхБэЯжГіИпЖШЕФЬивьадКЭбЁдёадЁЃДгжЧРћжЉжыжаЗжРыГіРДЕФыФЖОЫиHanatoxin-1ЃЌЬивьадЕизшЖЯСЫФЄЩЯЕФK +ЭЈЕРЁЃдкНсГІАЉЕФЗЂеЙЙ§ГЬжаЙлВьЕНСЫK +ЭЈЕРЕФИпБэДяЃЌOkadaЕШШЫЗЂЯжДгLachesana spжаДПЛЏЕФжТПзыФLaFr26. жЉжыЖОвКЖдЗЮАЉЯИАћЯЕ LX22 КЭ BEN гаЯИАћЖОадзїгУЃЌетСНжжЯИАћБэДяФкдДад K +ЕчСїЁЃAMPs дкжзСіжаЕФзїгУвВв§Ц№СЫШЫУЧЕФЙизЂЁЃвЛаЉ AMPs вбжЄУїОпгаПЙжзСіЛюадЃЌЖјСэвЛаЉдђДйНјжзСіЗЂеЙЁЃStrzelecka ЕШШЫКЯГЩЕФМђЛЏ ІШ-ЗРгљЫиРрЫЦЮядк 3D ХрбјФЃаЭжавжжЦСЫШщЯйАЉЯИАћЕФЩњГЄЃЌетБэУї ІШ-ЗРгљЫибмЩњЮяОпгаПЙАЉЧБСІЃЌПЙАЉыФЪЧбєРызгСНЧзЗжзгЃЌЭЈЙ§елЕўвРРЕадФЄЦЦСбгХЯШЩБЫРАЉЯИАћЁЃВЮПМПЙАЉыФЕФФЄЬивьадЯрЛЅзїгУЃЌAronson ЕШШЫжЦБИСЫвЛРраТаЭыФжЌжЪПХСЃЃЌЫќУЧПЩвдгыжзСіЯИАћФЄПьЫйШкКЯВЂНщЕМЯИАћЩБЩЫЃЌЖде§ГЃЯИАћМИКѕУЛгаЖОадЃЌБэУїСЫвЛжжаТЕФжзСіШмНтВпТдЁЃ
ЫфШЛаэЖрЖрыФдкСйДВЧАКЭСйДВбаОПжаЖМЯдЪОГіСМКУЕФПЙжзСіаЇЙћЃЌЕЋФПЧАжЛгаСНжжЖрыФБЛХњзМгУгкжЮСЦжзСіЃЌЗжБ№ЪЧгУгкЙЧШтСіЕФУзЗЈФЊЬцЕТКЭгУгкЖрЗЂадЙЧЫшСіЕФПЈЗЧзєУзЃЌЖјеыЖдЗЮАЉКЭЮИАЉЕШИќГЃМћжзСіЕФжЮСЦадЖрыФЕФжЮСЦВпТдбаОПШддкНјаажаЁЃвђДЫЙиМќдкгкбАевИќЖрдкжзСіЯИАћжаЬивьадБэДяЕФЪмЬхАаЕуЃЌВЂМгЧПЦфвНбЇзЊЛЏЁЃДЫЭтЃЌеыЖдИїжжжзСіЪмЬхЕФЖрыФзщКЯвВЪЧвЛИіЧБдкЕФВпТдЁЃ
ПЙВЁЖОыФ
ВЁЖОМФЩњгкЫљгаЩњЮяЃЌАќРЈШЫРрЁЂЖЏЮяЁЂжВЮяЁЂЯИОњКЭЙХЯИОњЁЃШЫРрвЛжББЅЪмВЁЖОадМВВЁжЎПрЃЌАќРЈАЃВЉРГібЊШШЁЂСїИаКЭЛёЕУадУтвпШБЯнзлКЯеїЃЈАЌзЬВЁЃЉЁЃОЁЙмЙ§ШЅЖўЪЎФъРДЃЌШЫУЧдкПЙВЁЖОвЉЮябаЗЂЗНУцИЖГіСЫОоДѓХЌСІЃЌЖржжПЙВЁЖОвЉЮявбЛёХњВЂЭЖШыСйДВЪЙгУЃЌЕЋАЌзЬВЁЕШвЛаЉМВВЁШдШЛУЛгагааЇЕФжЮСЦЗНЗЈЁЃ
ПЙВЁЖОыФЕФбаОПвбГЩЮЊвЛИіШШУХЛАЬтЃЌвђЮЊыФРрвЉЮяОпгаИпЖШЕФЬивьадКЭЛюадЁЃПЙВЁЖОыФжївЊЭЈЙ§АаЯђВЁЖОЛђЦфЫожїРДзшЖЯИаШОЁЃЖїЗђЮЄыФЪЧЕквЛИіЛёХњЕФПЙВЁЖОыФЃЌЪЧвЛжж 36 ИіАБЛљЫсЕФыФЃЌЫќЭЈЙ§НсКЯ gp41ЃЈHIV АќФЄЕААзЃЉЕФЦпыФжиИДНсЙЙгђРДзшжЙЦфШкКЯЃЌДгЖјзшЖЯ HIV ИаШОЁЃ2011 ФъЃЌПЙВЁЖОыФвЉЮяВЉШќЦУЮЌКЭЬиРЦУЮЌБЛХњзМгУгкСйДВжЮСЦБћаЭИЮбзВЁЖО (HCV) ЁЃЫќУЧОљгы HCV NS3/4A ЫПАБЫсЕААзУИНсКЯЃЌвжжЦЕААзУИЛюадЃЌДгЖјзшЖЯЫожїЬхФкЕФ HCV ИДжЦ. ИќЖрЙигкПЙВЁЖОыФКђбЁвЉЮяЕФбаОПе§дкНјааСйДВЧАКЭСйДВбаОПжаЃЌАќРЈеыЖд HBV КЭ HDV ЕФ myrcludex B ЃЌеыЖдСїИаВЁЖОЕФ flufirvitide ЃЌвдМАеыЖд HIV-1 ЕФ sifuvirtideЁЃ
2020ФъвдРДЃЌаТаЭЙкзДВЁЖОSARS-CoV-2в§Ц№ЕФКєЮќЕРДѓСїааадМВВЁбЯжиШХТвСЫШЋЪРНчШЫУЧЕФЩњЛюЁЃзд2020ФъГѕвпЧщПЊЪМвдРДЃЌПЦбЇМвУЧЭЖШыСЫДѓСПЕФОЋСІбаОПCOVID-19ЕФИаШОЛњжЦЃЌВЂбАевПЙCOVID-19ЕФжЮСЦЗНЗЈКЭвЉЮяЃЌАќРЈЖрыФРрвЉЮяЁЃCOVID - 19ЛљвђзщБЛПьЫйВтађЮЊвЛжжгаАќФЄЕФе§СДRNAЙкзДВЁЖОЃЌЛљвђзщДѓаЁдМЮЊ29.9kbЃЌгыђљђ№ЙкзДВЁЖОКЭSARS-CoVВЁЖОУмЧаЯрЙи
впУчБЛЦеБщШЯЮЊЪЧдЄЗРСїааВЁДЋВЅЕФгааЇЪжЖЮЃЌФПЧАвбдкаэЖрЙњМвЛёХњЪЙгУЃЌАќРЈЛљгкmRNAЕФвпУч ЁЂжизщЯйВЁЖОдиЬхвпУчЁЂУ№ЛювпУчЕШЁЃыФРрвпУчОпгаЬивьадИпЁЂАВШЋадКУЁЂвзЩњВњЕШгХЪЦЃЌГЩЮЊSARS-CoV-2впУчбаЗЂжавЛИіЛюдОЕФбаОПЗНЯђЁЃЛљгкИаШОЛњжЦЃЌЖрИібаОПаЁзщЩшМЦКЭЦРЙРСЫеыЖдSARS-CoV-2ЕФыФвпУчЁЃЫћУЧРћгУУтвпаХЯЂбЇММЪѕЗжЮіКЭЪЖБ№СЫЬивьадЪЖБ№SARS-CoV-2ДЬЭЛЬЧЕААзЕФBЯИАћКЭTЯИАћЙиМќБэЮЛЁЃLiЕШШЫКЭChakrabortyЕШШЫНЋЬьШЛБэЮЛађСазїЮЊеыЖдSARS-CoV-2ЕФвпУчКђбЁЮя ЁЃЖјBhattacharyaЕШШЫКЭWaqasдђГЂЪдЛљгкРДздBЯИАћКЭTЯИАћЕФБэЮЛЦЌЖЮЙЙНЈаТЕФыФзїЮЊCOVID-19впУчКђбЁЮя ЁЃЦфЫћбаОПаЁзщвВПЊеЙСЫРрЫЦЕФЙЄзїЁЃHerst ЕШШЫГЂЪдРћгУеыЖдАЃВЉРдњвСЖћВЁЖОЕФ CTL ыФвпУчбаОПЦНЬЈЩшМЦ COVID-19 ыФвпУчЃЌВЂЛёЕУСЫвЛЯЕСаыФвпУчКђбЁЮяЁЃаэЖрЦфЫћбаОПМЏжагкРћгУКЯГЩыФЛђКЫмеЫсзшЖЯ SARS-CoV-2 ЕФИаШОЙ§ГЬЁЃ
дк COVID-19 впЧщЦкМфЃЌПЙВЁЖОыФЕФПЊЗЂв§Ц№СЫЙуЗКЙизЂЃЌгШЦфЪЧеыЖд SARS-CoV-2 ЕФыФвпУчЕФПЊЗЂЁЃУтвпаХЯЂбЇБэеїЁЂЛљгкБэЮЛЕФЩшМЦЁЂМЦЫуЛњЪЖБ№КЭЗжзгЖдНгЕШаТММЪѕвбБЛбИЫйгУгкЩшМЦКЭЪЖБ№ыФвпУчКђбЁЮяЁЃОЁЙмЩаЮДгаыФвпУчЛёХњгУгкжЮСЦ COVID-19ЃЌЕЋдкыФвпУчЕФПЊЗЂЗНУцвбОЛ§РлСЫБІЙѓЕФОбщЃЌетаЉвпУчВЛНіеыЖд SARS-CoV-2ЃЌЖјЧвеыЖдЮДРДЕФаТВЁЖОЁЃ
НсТлгыЙлЕу
НќФъРДЃЌЖрыФвђЦфЖРЬиЕФЩњЛЏЬиадКЭжЮСЦЧБСІЖјГЩЮЊвЛРрЖРЬиЕФжЮСЦвЉЮяЁЃОЁЙмЖрыФдкФГаЉЗНУцгХгкаЁЗжзгКЭДѓЗжзгЩњЮяжЦМСЃЌЕЋгЩгкАБЛљЫсЕФФкдкОжЯоадЃЌЫќУЧЭљЭљДцдкФЄВЛЭЈЭИадКЭЬхФкЮШЖЈадВюЕФЮЪЬтЁЃЮЊСЫПЫЗўетаЉШБЕуЃЌШЫУЧЖдЖрыФвЉЮяЕФЗЂЯжЁЂЩњВњКЭгХЛЏНјааСЫЙуЗКЕФбаОПЁЃДЋЭГЕФЯШЕМыФЗЂЯжЗНЗЈгыКЯРэЩшМЦКЭЪЩОњЬхеЙЪОЕШаТММЪѕЕФНсКЯЃЌЮЊдкЖЬЪБМфФкПЊЗЂгааЇКЭбЁдёадЕФЯШЕМыФЬсЙЉСЫПЩППЕФЗНЗЈЁЃЛЏбЇКЭЩњЮяжизщКЯГЩЗНЗЈЕФЕЅЖРЛђСЊКЯЪЙгУПЩвдИпаЇЁЂПЩППЕиДѓЙцФЃЩњВњКЯГЩыФЁЃетаЉыФПЩвдЭЈЙ§ЛЏбЇКЯГЩЛђвХДЋУмТыРЉеЙвдЮЛЕуЬивьадЗНЪННјвЛВНаоЪЮЃЌвддіЧПЦфЮШЖЈадКЭЩњРэЛюадЁЃ
ЫфШЛжЮСЦадыФСьгђЪМгкЬьШЛМЄЫиЃЌЕЋДЫКѓЗЂЯжКЭПЊЗЂЧїЪЦвбДгМђЕЅФЃЗТЬьШЛМЄЫиЛђРДздздШЛНчЕФыФзЊБфЮЊКЯРэЩшМЦОпгаРэЯыЩњЛЏКЭЩњРэЛюадЕФыФЁЃЗжзгЩњЮябЇЁЂыФЛЏбЇКЭыФЕнЫЭММЪѕЕФжиДѓЭЛЦЦЪЙыФвЉЮяЗЂЯжЁЂыФЩњВњМАЦфжЮСЦгІгУСьгђШЁЕУСЫжиДѓНјеЙЁЃЦљНёЮЊжЙЃЌвбга 80 ЖржжжЮСЦадыФНјШыШЋЧђЪаГЁЃЌЪ§АйжжыФе§дкНјааСйДВЧАбаОПКЭСйДВПЊЗЂЁЃетаЉыФвЉЮявбБЛгІгУгкЙуЗКЕФМВВЁЃЌР§ШчЬЧФђВЁЁЂаФбЊЙмМВВЁЁЂЮИГІЕРМВВЁЁЂАЉжЂЁЂДЋШОВЁЁЂвдМАвпУчПЊЗЂЁЃПМТЧЕНЦфОоДѓЕФжЮСЦЧБСІЁЂЪаГЁЧАОАКЭОМУМлжЕЃЌЮвУЧдЄМЦжЮСЦадыФНЋМЬајЮќв§ЭЖзЪКЭбаОПСІЖШЃЌВЂШЁЕУГЄЦкГЩЙІЁЃ
Утд№ЩљУїЃКБОЮФЮЊаавЕНЛСїбЇЯАЃЌАцШЈЙщдзїепМАддгжОЫљгаЃЌШчгаЧжШЈЃЌПЩСЊЯЕЩОГ§ЁЃЮФеТБъзЂгазїепМАЮФеТГіДІЃЌШчашдФЖСдЮФМАВЮПМЮФЯзЃЌПЩдФЖСддгжОЁЃ