
ժҪ �Ի��Ľ����˷���, ���Ծߴ����Եľ����͡�DKP (Diketopiperazine, ��ຶ�ͪ)�͡������͡�ϩ���͡����������͡��������͡���(��)���͡�Mannich ���͡������ͼ� Freidinger �ͻ��ĵĺϳɷ�����һ����������.
����ʮ��������Ȼ�硢�����ڻ�ѧ�ϳɵõ��Ĵ����Ļ�����, �Ը�������ԡ��Ͷ������õ��ص�������ҩ�ﻯѧ�ҵĸ߶ȹ�ע. ���������, ֱ���ĵķ���������(flexibility)��ɵĹ����ױ�ʹ���������ϵ�ǿ�ȼ�ѡ�����ܵ�Ӱ��. ����, �����ڵİ���ø������øҲ�ܷ���ش�ֱ���������˻����и�����, ʹ֮����. ��������Ļ�������, ʹ�乹����(constrained conformation)�Ǹ����ķ��������ȶ��ԡ����������Ե�����;��֮һ. �����о��Ѿ�֤��, ��ֱ�����ȵ��ṹ�ĸ�Ϊ���ĺ�ʹԭ�е�����������ʮ����������. ������п������������������������ߵ��ڵȻ��Ե���Ȼ�������������в�ͬ���͵����������ṹ. ������ӻ��ĵ�ϵͳ��������, ��һЩ����(loop)�в�ͬ�����ṹ�Ļ����������ϳɷ�����������.
1 ���ķ���
��ζ�����Ȼ��������Ҫ���õĻ��Ľ��з���, ��������һ��Ȩ���ġ��ϸ�ı�. ���Ĵ����¼�����ͬ�ĽǶȽ��й���:
1.1 ���������Ƕȷ���
���������Ƕȿɽ����ķ�Ϊ������(Homodetic cyclopeptides)���ӻ���(Heterodetic cyclopeptides)����. ������ָ��������������������, ��������ṹ��һ; �ӻ���ָ�������г�����������, �������������ṹ, ����ְ�����������.
1.2 ����Դ����
����Դ�Ƕȿɰ�ͼ 1 �Ի��Ľ��з���:
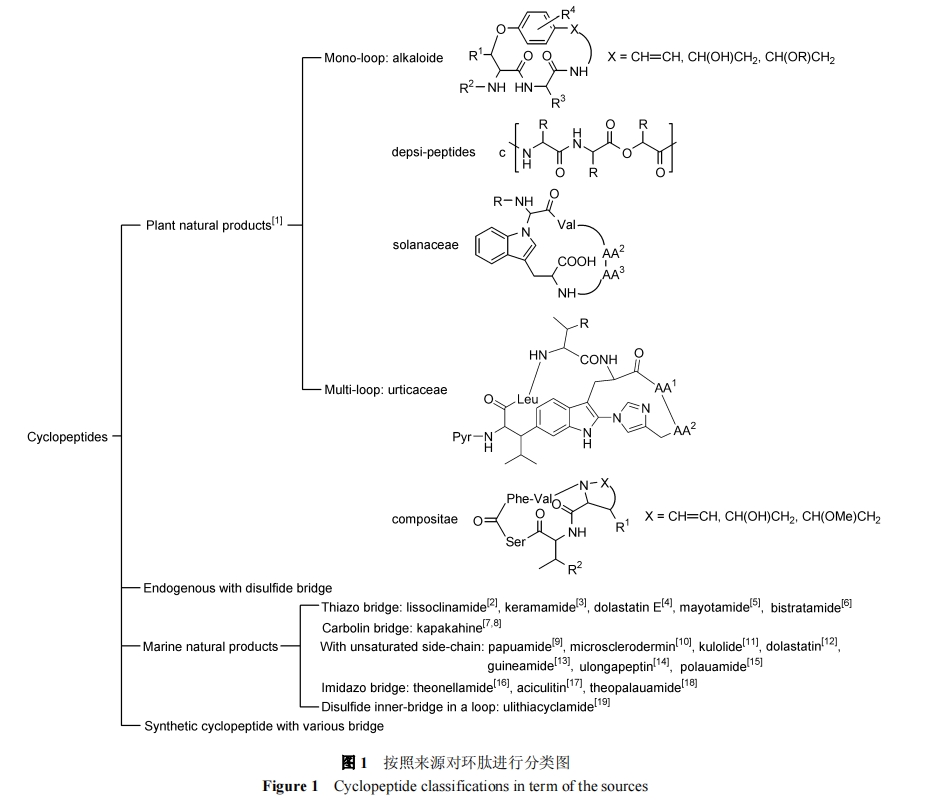
1.3 ����ͷλ�÷���
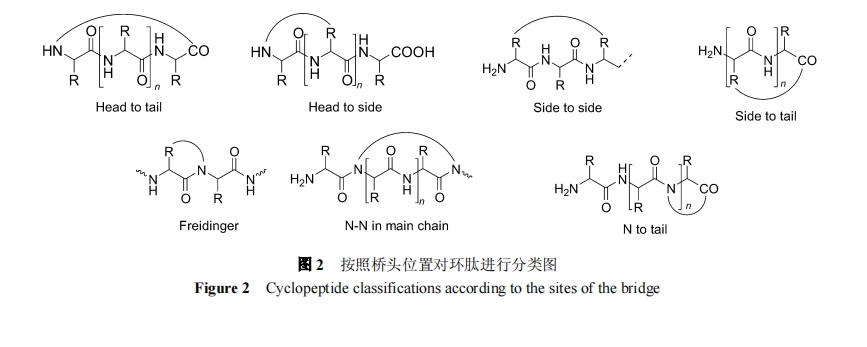
����ͷλ�ÿɽ����ķ�Ϊ 7 ��, ��ͼ 2.
1.4 �������ṹ����
�������ṹ�ɽ����ķ�Ϊ���价�ĺͷǾ��价�ľ��价���ְ����������š������źͶ�����; �Ǿ��价�İ������š�ϩ�š����������š�����֧���š����š������š�Mannich ���š�PNA (Peptide nucleic acid, �ĺ���)�ŵ�.
1.5 ��������������
���������������ķ�Ϊ�� loop ��, ˫ loop �ͺͶ�loop ��.
2 ���价�ĺϳ�
���еĸ��ֻ��Ļ�������, ��������(�ֳƾ�����)���������������(depsi-)Ϊ�����ṹ�Ļ���ռ�������, �������ǵĺϳɷ����Ƚϳ���������. ��������ֻ����ֱ���Ϊ���价��.
2.1 ������Ϊ�ŵĻ��ĺϳ�
����ĵĺϳ����ܶ�, �����Ͽ��Դ���������ʽ: (1)ʹ�����ϼ����Ȼ����; (2)�����ϼ��ķ����ڰ�����. ǰ�߶Ծ�����Һ�������෨������, ���߶����ڹ���ϳ�. ���������ص����������ĺϳɷdz���, ���������һЩ�д����Եķ���.
2.1.1 �������ӹ��෨
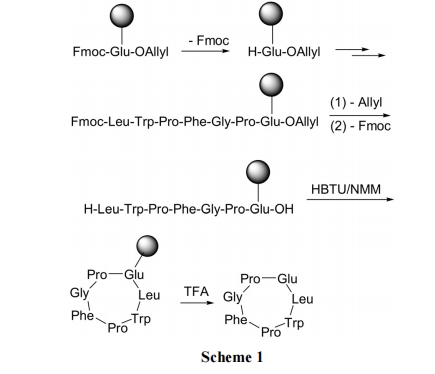
���ùȰ���(���춬����)�IJ���COOH����������γ���������. ������ѳ� Fmoc, ʹ�Ȱ���� ��-��������, �Ա��� N ����װ����. ȫ��������ɺ�, �����Լ��ѳ� C �� Allyl, ʹ�Ȱ�����Ȼ�����; ���ѳ� N �˵�Fmoc, ʹ����N�˰�������. ��������ϼ�HBTU������, N �˼� C �������γ����������γɻ���(Scheme 1)[20].
2.1.2 ��Һ��ϡ�ͷ�
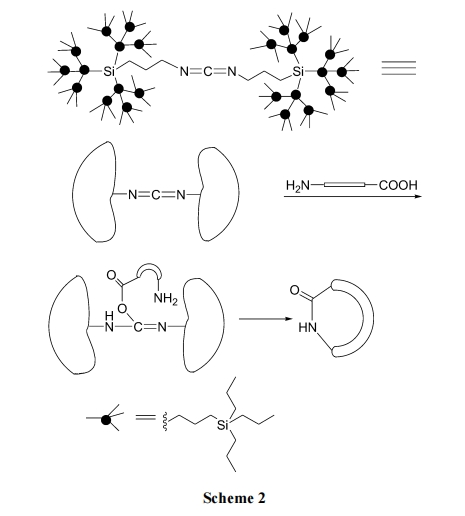
�ڴ�ͳ��Һ�����з����ڻ��Ϸ�Ӧʱ, Ϊ�˱�����Ӽ�����ϱ���ʹ��Ӧ������Һ�о���ϡ��. Ȼ�����ָ߶�ϡ�͵���������ɷ�Ӧʱ���ӳ�������Ӧ�϶�, Ҳ����������������. ����������, Amore ��[21]ʹ��һ�ֿռ�λ������ͻ״����ȡ����̼���ǰ���Ϊ���� �� , �� �� �� �� �� �� DCC (N,N-dicyclohexylcarbodiimide, ��������̼���ǰ�)������Һ��ʽ�����������ĺϳ�, �õ����Ⱥܸߵ�Ŀ�����(Scheme 2).
2.1.3 ��������ڰ��ⷨ
�����������(RCOON��R')�������Լ��߶�����, ����ʹ�����Ϊ linker �Ĺ�������, �Ʊ�ͷ-β���ϵ�����������. ���һ�����ϲ������ϼ�����ͬʱ��ɷ����ڰ��⼰�ѳ���������(Scheme 3)[22].
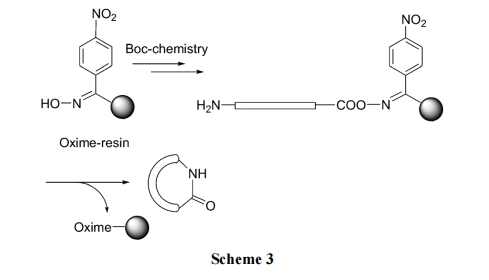
2.1.4 ���ౣ�������ͷ����ڰ��ⷨ
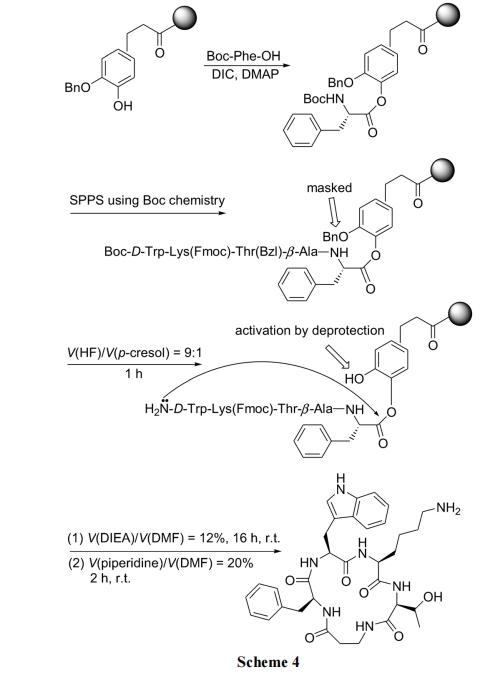
��������(Safety-catch linker)���ص�����Ŀ��ṹ�ĺϳ��зdz��ȶ�. ֻ�о��ض��Ľṹת��, �� linker�����ŷ����ѽⷴӦ[23]. ����Ļ��ĺϳ�������������Ϊ������֬ linker �ṹ, ��������װ��ɺ��ѳ�linker �ϵ��л�, ʹ��λ�ķ������. ��ʱ���� N �˰�����������������, ���������ڰ���, �õ����IJ���(Scheme 4)[24].
2.1.5 ��������������/�����ڰ��ⷨ
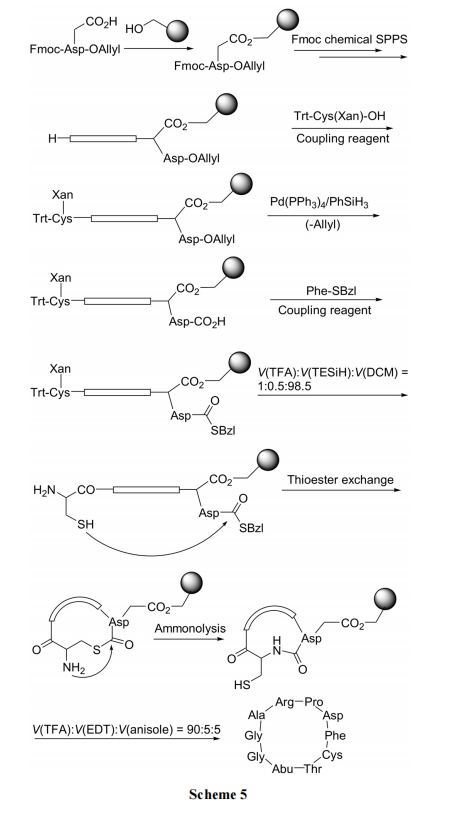
��Ҫ�ϳɲ�������Ϊ: (1)ʹ Fmoc-Asp-OAllyl �IJ��� COOH ���ǻ���֬����Ϊ������ linker; (2)�ѳ�Fmoc, ���� N ����װ����; (3)�� N ������ Trt-Cys(Xan)��Ϊ�������������ⷴӦ��ǰ��ṹ; (4)�ѳ� C ��Asp-OAllyl �ϵ� Allyl ʹ COOH ��������� Phe-SBzl����, �γ�����������Ӧ�ĵ���ṹ; (5)�ѳ� N �� Cys�ϵ�����������, ʹ ��-NH2�� SH ����; (6)�� pH 7.5 �����½��з������������������ⷴӦ, �γ�����������; (7) TFA �г� Asp �����ϵĹ�������, ʹ��������, �����ʸߴ� 80% (Scheme 5)[25].
2.1.6 α Pro Э�����Ļ�����
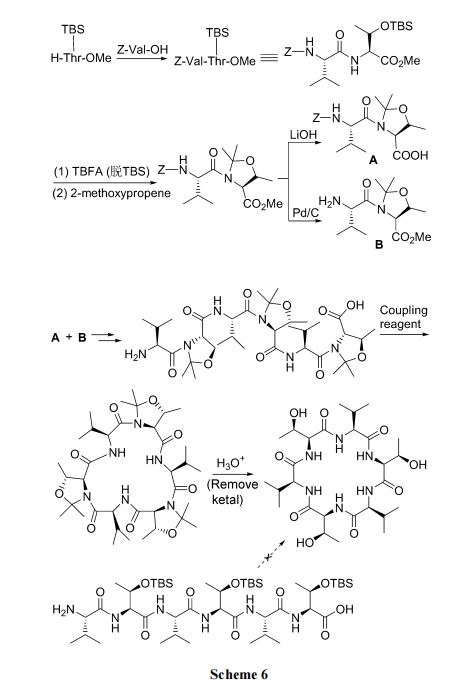
���� Pro �� Gly �Ķ�����������Ӱ����ѱ�����. Ϊ�˿�������װ����ʱ��Ŀ�������е� Thr �л��ñ�ͪ����, ��ʱ������Ԫ��, ʹ��ģ�� Pro ��ת���շ��ṹ(turn inducer), �ﵽ�������������������ڻ�����Ŀ��. �������γɺ�, ���º��ᴦ���ѳ���ͪ��������, �ָ�Thr �Ľṹ. �˷�ʽʹ�����������ɣ�5%��ߵ� 80%����(Scheme 6)[26].
2.2 �����Ż��ĺϳ�
���ֻ��ĺϳɵķ���ѧҲ�ܳ���. ��Ҫ�ϳɷ�ʽΪ����װ����, Ȼ���ѳ����װ��ἰ�����л��ϵIJ���������. ������ʵ�����������, �����, H2O2, I2, Hg2����, Fe3����, DMSO �Ƚ������ SH ����Ϊ�����. Ӧ��ע���������Һ�н��з�����������Ӧʱ������������ĵ�Ũ�Ȳ��˹���, һ���� 1 mmol/L֮��. �������ڷ��Ӽ���ŵĸ���Ӧ. ����DZ�֤�����ڳɼ��ĸ�ϡ��ԭ��. ���֮��, ����ϳɷ��ӵļ�ϡ��ЧӦ(Pseudo-dilution effect)���Ի�����֤��������������[27]. ��Ŀ��ṹ�к����������϶����ʱ, �����ȷ���ű��Ϊ�ؼ�. ���潫���ܼ��ֲ�ͬ�ĺϳɲ���, �����ȷ�γɶ����(���� Folding)������.
2.2.1 ����ѧͬ���ϻ���
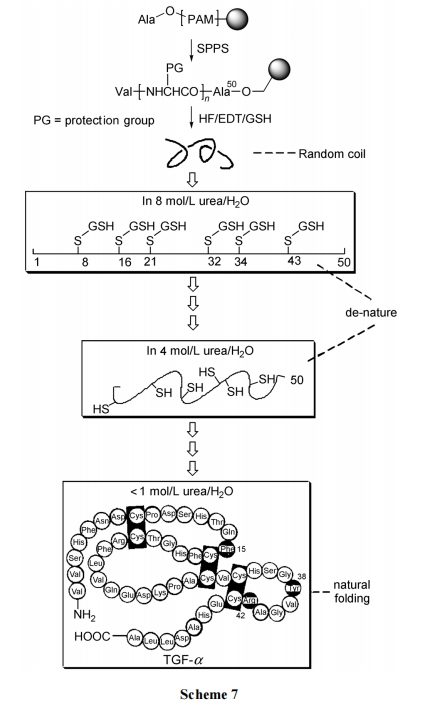
�Թ����ĺϳɷ�ʽ�ϳ�ת����������(TGF)-��(��50 ���л���6 �����װ���)Ϊ��. ���� 6 �����װ���IJ����ϻ����� MBzl(�Լ���)����. ���һ���� HF �ѽ�, ���г���֬��ͬʱҲ�Ѱ��� MBzl ���ڵ�ȫ�����������ѳ�. ������ HF �ѽ��Լ��м��������Ļ�ԭ������(GSH), ��Ч����ֹ�� TGF-�� ������ 6 �� Cys �л�����������. Ȼ��ʹ�����͵Ĵֲ�Ʒ���ڸ�Ũ��(8 mmol/mL)������ˮ��Һ��, ��ʹ TGF-�� ��������չ��, �ʡ����ԡ�(de-nature)״̬. �ٽ�����Һ����������, ���Ŵ�����Һ����Ũ������, ʹ���̬�� GSH ���������ͬʱ, TGF-�� ��Ҳ��ԭ������չʽ de-nature״̬����Ȼ����. ���������Դ�Ե�TGF-�����еľ���״̬(�����Զ�����γ�)Ӧ������Ȼ�γɵ��������״ ̬ . �о���֤�� , TGF-�� �����Զ��������Cys8-Cys21, Cys16-Cys32, Cys34-Cys43 ����ʽ���ڵ�. ���, ��ʱ�䡢�����ؽ�����ȷ�Ķ����������һ������ѧ���ƹ���. ���������Ӧ������ǿ��ʱ�����, ���ܳ��ִ�����ŵ����. ���ֲ���ȷ�� Folding �ᵼ��������Ե�ɥʧ(Scheme 7)
2.2.2 ��������/�ֲ��ϻ���
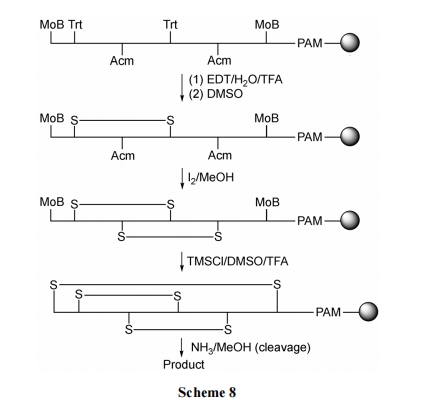
���ö��ѳ���Ӧ�����Բ�ͬ�ı�����, �ֱ𱣻���ͬλ�� Cys �ϵIJ����ϻ�, ʹ���� Cys �ڲ�ͬ��ѧ�����зֽν���ѡ������Դ���, ����������/�ֲ��ϻ��Ļ���ԭ��. ���ַ�ʽ��ʵʩӦע�����¼���: (1)��ΪҪ������������ɻ���Ӧ, Ϊ�������ٷ��Ӽ���ŵĻ���, Ӧ�Թ���Ϸ�ʽΪ��. (2)��Ҫ������һ�� Cys ������ͬ�ֱ�����. (3)���� Cys �IJ�������������������Cys �ı�������ͬ, �������ǵ��ѳ���Ӧ�����Բ����������, �Ա㻥������, ��������ЧӦ. (4)ȫ��(���һ�� Cys ����)Cys ���������ѳ���Ӧ��Ӧ��ɹ��������� linker ������ǰ�ѽ�. Scheme 8 �����˺����Զ�������ĺϳ����, ����һ�����������е� Trt(������)����; �ڶ�����ǿ�����е� MoB(�Լ�������)����. ����֮���������ݶȲ����з�ʽ. �������� Acm ����, ����ǰ���ֱ�������Ϊ�������з�ʽ.
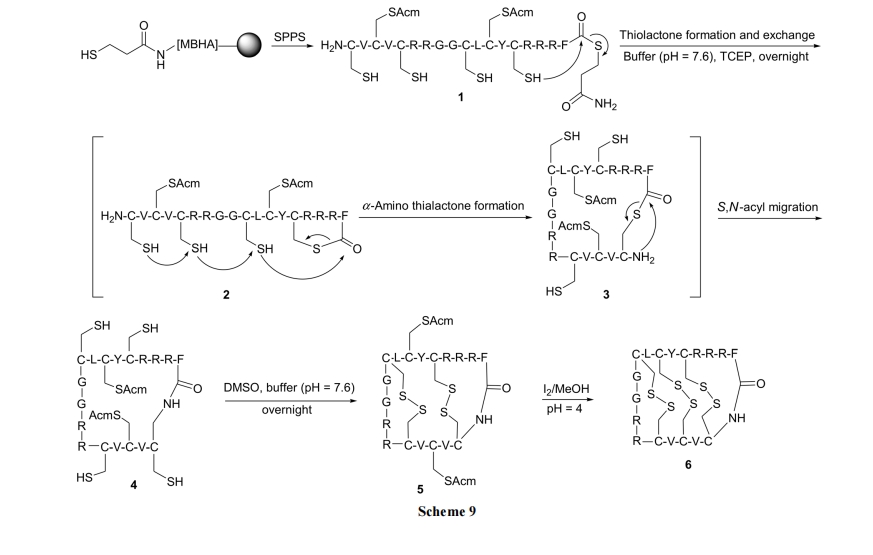
2.2.3 ����ŵ����������
Tam ����[28]������һ�ַ��㡢���С�������Ʊ����ض����Ż��ĵĺϳɷ���. ����Ŀ�껷�ĵ�ǰ�������N �˾���һ�� Cys �л�, �� C �˺��������ṹ, �����������м���躬��һ�������ϵ� Cys �л�. ���м��� 1, �� pH Ϊ 7.6 ��ˮ��Һ��, ���ȷ���������ӽ� C �˵�SH����C��������������������Ӧ�õ��м���2. ������� C �� N ����Ķ������������Ӧ, ��������(�ֳƶ���ŵ)ЧӦ, ֱ�� N �����һ�� SH ��Ϊ����. ��Ȼ, �м��Cys�л��ָֻ�������SH״̬��Ϊ�м���3. ��ʱ������ N ���� C ������, ��� N �� Cys �� NH2���������һ���������з����ڰ��ⷴӦ, �������������� 4. �� 4 ��, ���ڻ�������Ӱ��, �������ڵ� SH �����γɶ����. ��λ����Խ�Զ������� SH ֮��������γ�����ѧ�������Ķ�����(�� 5)�Ľṹ. ���һ�Դ��� Acm �����������ϻ����ѳ� Acm ��, Ҳ��Ϊ������, �õ��� loop �ͻ��IJ��� 6 (Scheme 9).
2.3 ������(Cyclo-depsipeptides)�ϳ�
2.3.1 Kahalalide B �ĺϳ�
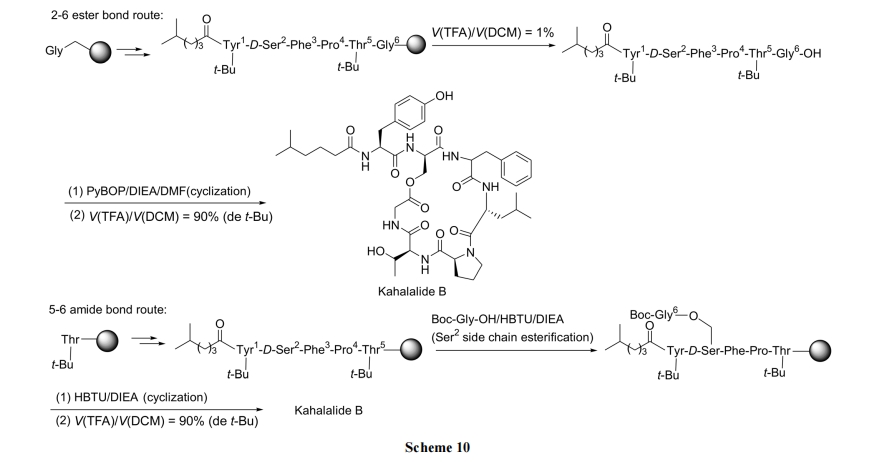
����Ȼ���ﻷ���� Kahalalide B �ĺϳ��в��������ֺϳ�·��, ���õ���Ŀ�����(Scheme 10)[29].
2.3.2 Kahalalide A �ĺϳ�
�������� Kahalalide B ����, ����Ŀ�껯����Ҳ�Ǻ�һ���������Ļ���[30]. ���������ϳɷ���������Ƴ��ȹ�����������ֱ����, ���������������ʽ�ػ�(Scheme 11).
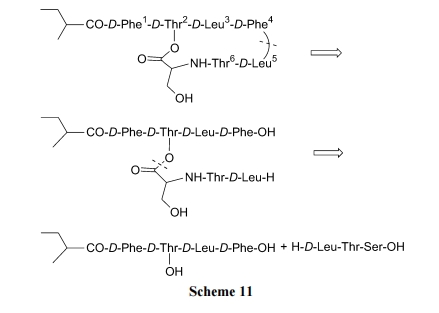
���ϲ�ͬ����, ���ϳɲ��ù��ౣ���� linker ����, �Ƚ�Phe4���������֬����, ���������ϵõ������м��� 7. �ѳ� 7 �� Thr ���� t-Bu ��ʹ OH ����, �Ա��� Ser7��������. ����ٽ��� Thr6�� D-Leu5, �õ�ֱ����ȫ���нṹ 8. �����ѳ� Fmoc ���Լ�������ऻ���Ż��Ļ����� linker �� D-Leu5 �İ���֮��İ���(���һ��)��Ӧ, ����Ҫ�ڻ��������֮ǰ��ʹ D-Leu5 �ϵ�Fmoc ת��Ϊ Trt, ��Ϊ�м��� 9. Ȼ���� ICH2CN ���������� N ԭ�������, ʹ���� linker �. ���� TFA �ѳ�N �˵� Trt, �û���ǰ�� 10. ����� DIEA �����·��������ڰ��� , ͬ ʱ �� �� �� ֬ �õ����� Kahalalide A (Scheme 12).
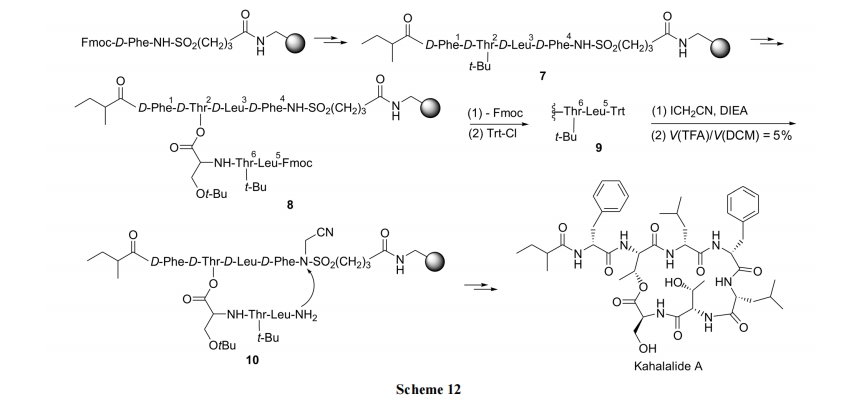
2.3.3 �Զ��յ��� AIP �ĺϳ�
�����ϳɵ�Ŀ�껯���� AIP (auto-inducing peptide, �Զ��յ���)Ϊ��-β�������ɻ��Ļ�����. �úϳɲ��Ե��ص���: (1)������Ϊ�����ѽ��� linker; (2)�� linker �����ȼ��ϵIJ������� C ĩ�˵ĵ�һ���л�, ���ǵڶ����л�; (3) C �˵�һ���л��������м� Ser �л��IJ���OH ����������. ��ƴ��ֺϳɷ�ʽ������������������ linker �����ĺϳ��еķ�Ӧ���������Ժ�ǿ, �������ᡢ�����Ҳ���ȶ�. Ȼ������ linker �ṹ����������, �� Cu2��, I2, NBS �ȷdz�����, �����ܵ��˻��ŵĽ������ѽ�. ���, Ser �����ϲл��� NH2���Է���ض����� linker ���з����� SN2 ��Ӧ, �γ�������, ͬʱ�г���֬(Scheme 13)[31].
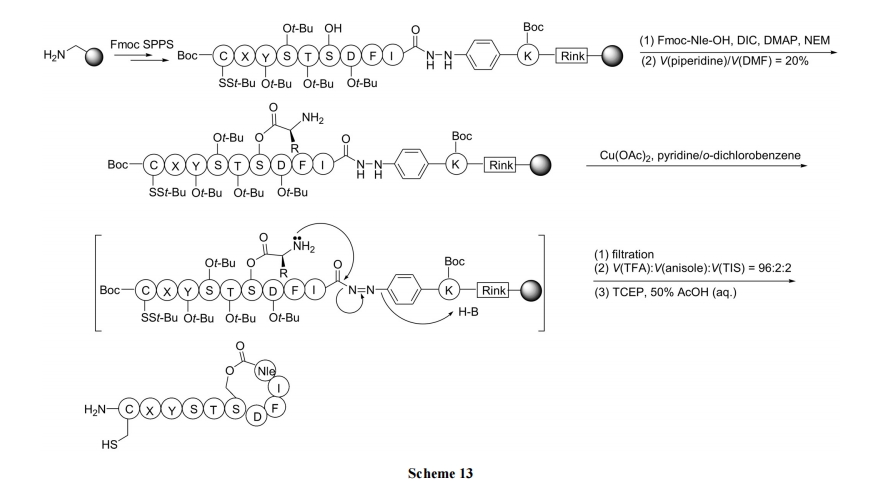
2.3.4 �f��ͪ��������
Heimgartner ��[32]�����һ����Ϊ��ֱ����������(direct amide cyclization)���ĺϳ�·��, �Ʊ����� ��,��-˫ȡ���л���ɵ�������. ��ԭ����ֱ���� C �˵Ķ��������ṹ���� ˮ HCl �� �� �� , ������һ����azirine/oxazolone������̬ 11 �� 12. �����ܵ�ͬ������N ���ǻ��Ľ���, ʹ oxazolone ��Ԫ������, ͬʱ�� OH�γɷ���������(Scheme 14).
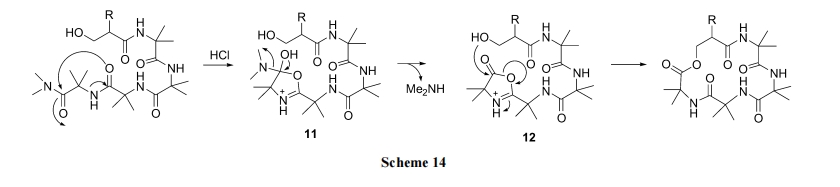
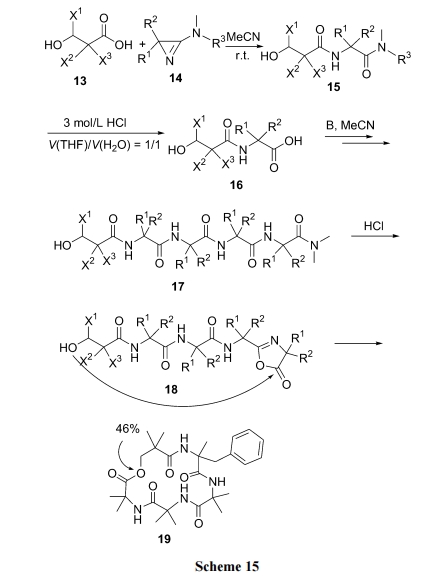
���п��Կ���, ��,��-˫ȡ��������Ľ����Ƿdz���Ҫ��, ����ȷ�������� N ���ǻ��� C �˵Ľӽ�, �����������γ�. Koch ��[33]���ô˷�ʽ�ϳ���һЩ������ 19, �������ʴ� 46% (Scheme 15).
2.3.5 Ugi ��Ӧ������
��ȩ�����������������������(���ֹ��ܻ�)Ϊ������е� Ugi ��Ӧ, Ҳ���Եõ���һ�������������IJ���. ����, ���� CHO, NC, NHR ���ֹ��ܻ�����, ���ɇf���м��� 20. �پ��������������շ�������, ��������˫�f���м��� 21, ������ɻ�����(Scheme 16) [34].

3 DKP ����������ϳ�
3.1 �������ķ����ڰ���
�����Ǿ�����Һ���Ʊ��Ķ�����, ����������Ϊlinker �Ĺ��������ϵĶ���, �� N �˵ı��������ѳ���, ������� HOAc �Ĵ��º����������� NH2 �Է����ڵ� C ĩ�������İ��ⷴӦ(Scheme 17).
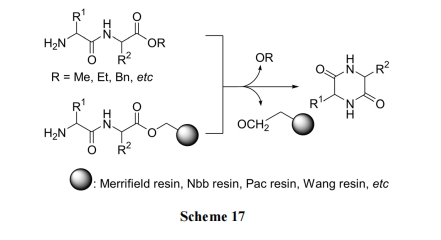
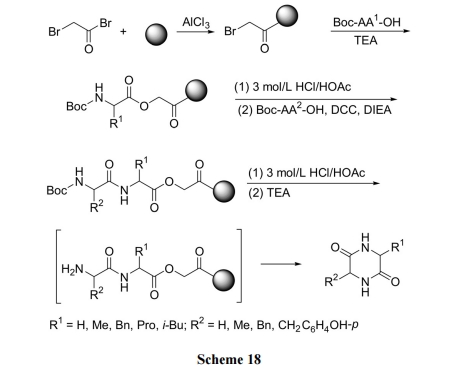
ֵ��ָ������������Ӧ�ھ������������Ϊ linker�Ĺ����ĺϳ�����һ���ɵ���(�������ϵ�)ȫ�ϳ�ʧ�ܵ����ظ���Ӧ. ����[35]���������������Ӧ, �� Pac ��֬Ϊ�����Ʊ��˶������������ DKP �����IJ���(Scheme 18)
3.2 BAL ��֬Ϊ����Ķ��ķ����ڻ���
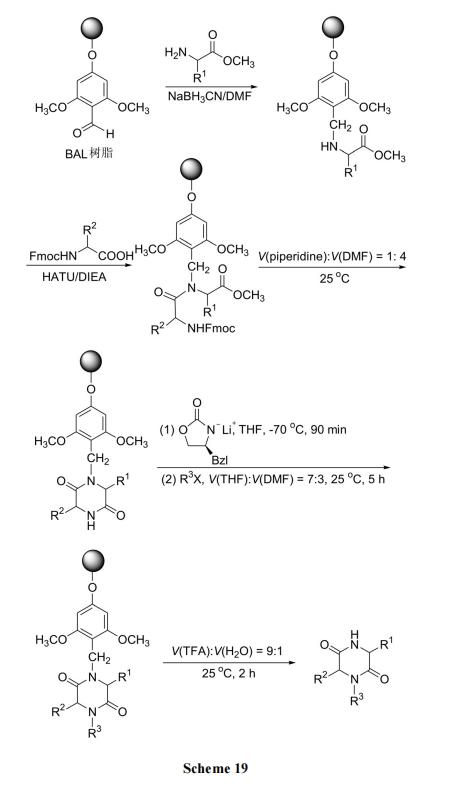
�������� DKP �ķ�ʽһ����� DKP ǰ����Ȼ�����֬�� linker ��������ʽֱ������, ��˰��������������������ڻ���ʱ�γ� DKP ��ͬʱ����֬���и�. ���� BAL ��֬Ϊ����� DKP �ϳ���ͨ�� linker �� DKPǰ��İ�������, ���������γ� DKP, �������֬�и�, ��������Ӧ�ֿ�����. ����, ������ DKP ��, ͨ��ϴ�ӳ�ȥ��Һ�е�����, �и��õ��ߴ��ȵIJ���[36](Scheme 19).
3.3 ȩ�백����Ļ�ԭ�����;���ϳ� DKP ������
���� Wang ��֬�ϵĵ�һ��������� NH2����Ӧ��ȩ�����ϳ� Schiff ��, �����ڻ�ԭ�� Na(OAc)3BH ������, ����N������м���22. �������һ��Boc-������� 22 ���ٰ����������� 23. �ѳ����ߵ� Boc �����ڼױ��м���, ���������ڰ��Ⲣͬʱ�г���֬, �õ� N ����ȡ���� DKP ����[37] (Scheme 20). ��������� Na(OAc)3BH ��, ������ʹ�� NaCNBH3 Ϊ��ԭ��, ����������� PAM ��֬����ϰ����Wang ��֬, �õ������ʸ�����IJ���[38].
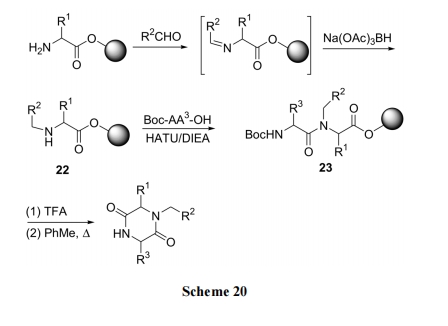
3.4 N,N'-˫ȡ�� DKP ������ĺϳ�
�� 2-�����������ȶ���֬�ϵ�һ��������� NH2�� �� �� ʱ���� , ʣ ��һ�� H ԭ �� ȷ �� �� �� �� ��Mitsunobu ��Ӧ�õ� N ��ȡ������ 24. �ѳ� N �ϻ��������ٱ���������, ���м���25. ��SN1��Ӧ��25ת��ΪN ȡ���ĸʰ������� 26. ������ TFA-H2O ��������ֱ������ 27 �� DKP ������ 28 �Ļ����. ������������(TFAA)/DCM��������, 27����ȫת��ΪĿ�����28[39](Scheme 21).
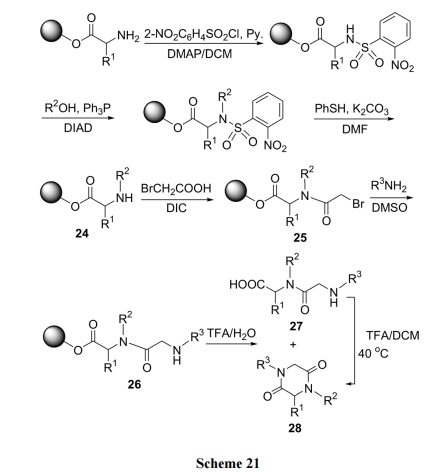
3.5 Ugi ��Ӧ;���ϳ� DKP ������
Ugi ��Ӧ����ȩ���ᡢ�������� 4 �ַ�Ӧ������ж����һ����Ӧ(MCR). ���ڷ�Ӧ�������Ͷ�, ��˲�����кܺõĽṹ������. �����ǹ��෴Ӧ��ʽ, ���Լ�Ӧ��Ĵ�������, �����˹��ڹ��������ϵ����ϲ�����, ����ʣ��ļ��ַ�Ӧ����ɹ��˳�ȥ[40](Scheme 22).
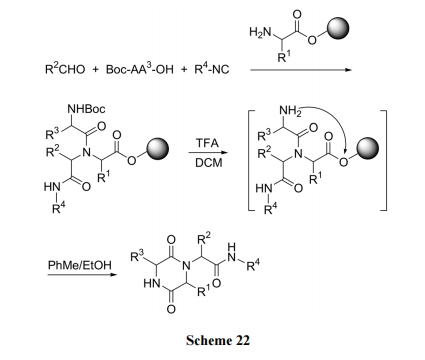
3.6 ���� DKP ������ĺϳ�
������Ե� DKP ����Ȼ���P�˹���ƺϳɵ�DKP ������ĽṹԶ�������ϳ����IJ���Ҫ���ӵö�, �����������в����ĹǼܽṹ. �������ĺϳ�����һЩ���ױ���[41,42]. ����ĺϳ��Ǹ��������Ԫ��Ϊ�Ʊ� DKP ����ǰ��֧�ܽṹ. ���Ƚ����˱������Ǹ����� Fmoc-Hyp-OMe �IJ��� OH �� Ellman ��֬��˫�������ӳɷ�Ӧ��������. ���ʹ Hyp �л��� ��-̼�����, ���õڶ��ְ������� Hyp �ӳɶ���. �ѳ��ڶ�λ�л���N ������, ʹ���� NH2���� Hyp �ļ�����, ���������ڰ���, ���� DKP �����ṹ(Scheme 23) [42].
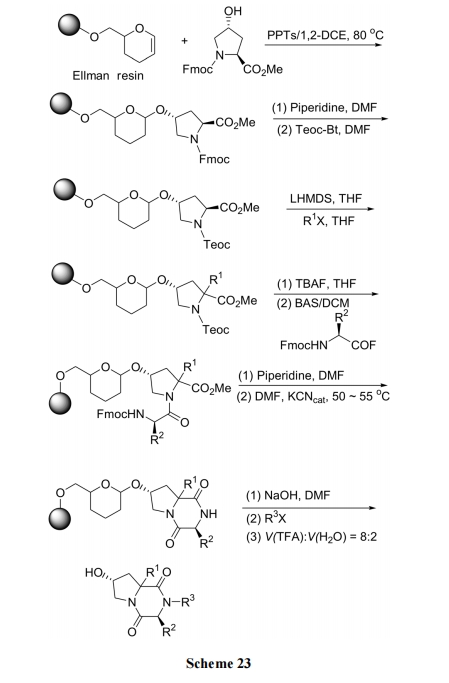
3.7 ϩ���� DKP �ĺϳ�
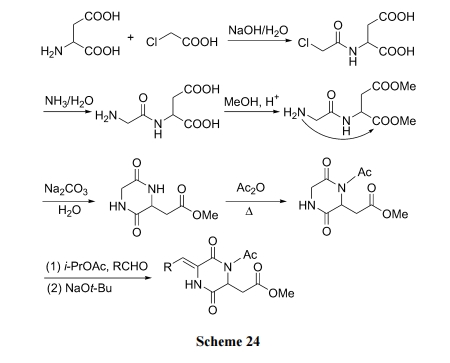
���������۵���������������춬��������, ���ð�ˮ���ȷ��� SN2 ��Ӧ�Ƶ� Gly-Asp ����. ���������Լ״�������ʹ Asp ���Ȼ�������, ������ڰ������� DKP �Ͳ���. ȫ���ϳɵ�����ص��Dz��þ�����������Լ�, Ҳû���κα������ѱ����ķ�Ӧ, ��˳ɱ��͡����ڴ�����[43] (Scheme 24).
3.8 ��˫�������Ϸ�
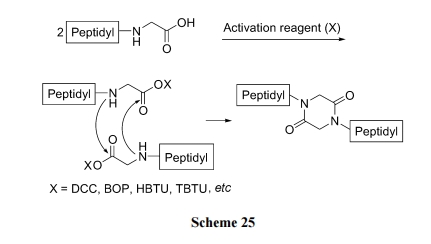
ǰ����ܵĸ��ֺϳ���, һ���Ҫ�������ķ����ڰ����γ� DKP ��;��. ��ʵ����Ϊ�����˫��������Ҳ��һ�ֺϳ� DKP �ṹ�ĺ�����ʽ. ��Ϊ���ַ�ʽ�õ��� DKP ��������� N,N'-˫ȡ���Ľṹ�ص�. ���ֺϳ���Ҫ��Ӧ��� C ĩ�˺��� Gly �л�, ��Ϊ�����л��Ħ�-̼ԭ���Ͼ��в���ȡ��, λ��Ӵ�����˫���ӷ�Ӧ. �о�����, ������ľ�˫���������γɵ�DKP˫�������ṹ��, ��ԭ�еĻ��Կɻ�����. ��˺�DKP���� ˫ �� �Ļ������ �� �� ��ϳɾ���һ�� �� �� [44](Scheme 25).
3.9 �ݻ� DKP �����ĵĺϳ�
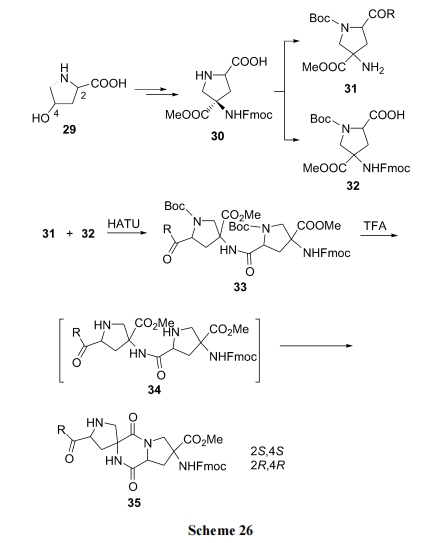
���Ǹ�����(29)Ϊԭ��, ���ಽת�����ɹؼ��м��� 30, �ٽ� 30 �ֱ�ת��Ϊ������� 31 ���Ȼ���� 32. �����������Ϊ���� 33 ֮��, �ѳ��� 32 �����Ľṹ���ϵ� Boc, ʹ�ٰ�������� DKP ǰ��ṹ 34. ����ڼ�������, �������ķ����ڰ���, ����һ����Ԫ����DKP ��Ϊ�ݻ��IJ��� 35. ������ݻ� DKP �������е�һ����Ԫ�ṹ[45] (Scheme 26).
4 �����Ż��ĺϳ�
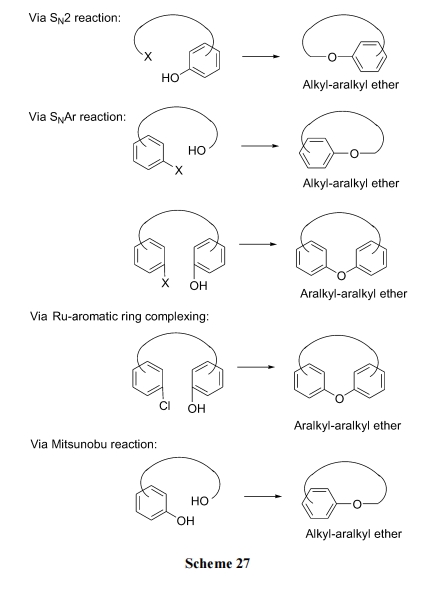
�������ṹ��, �ѻ����ַ�Ϊ֬-�������ͼ�������������, ����ǰ��Ϊ����. �������ŵ����ɷ�ʽ, ��Scheme 27 ��ʾ��Ӧ;��.
�ںϳɲ�����������������, �ѻ��ĵĺϳ�˳����Ҳ�����������: (1)���Ʊ���������ṹ, ������Ѽ�������ʵ�ֺϻ�. (2)���Ʊ����ѽṹ��Ƭ��, �����������(��������)������ʵ�ֺϻ�.
4.1 ���ѻ��ĺϳ�
��Ȼ�����ѻ��� Sanjoinine G1 �ĹǼ��Ϻ��������ļ���һ������/��������Ѽ�. ����ϳɷ������ʮ��Ԫ����Ŀ��ṹʱ, ����ȷ�������ɻ���λ�� a �� b (Scheme 28). ��˴�������·�߿�����ɴ˻��ĵĺϳ�.
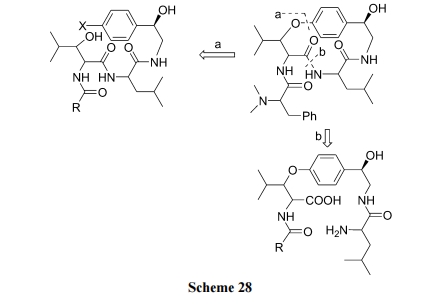
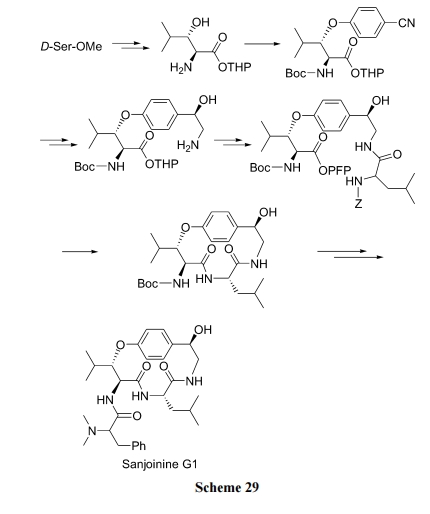
·��(a), ���Ʊ����л��ѽṹ��ֱ����, ��������������ʽ�ػ�(Scheme 29)[46].
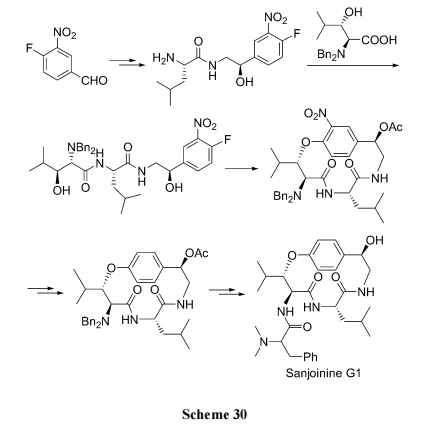
·��(b), ���Ʊ�����, ��� SNAr ��Ӧʹ������������ϵ��ٴ�֮����һ���� HF ���ɻ��Ѽ�(Scheme 30)
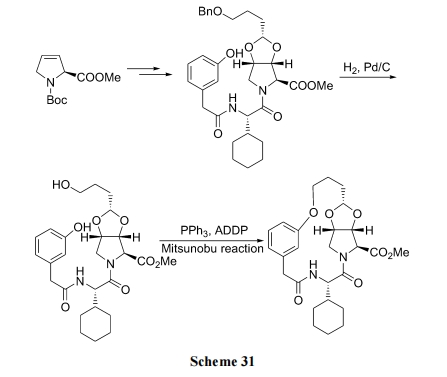
���� Mitsunobu ��Ӧ�Ʊ� ���ѻ�����(Scheme 31)[47].
���������� Tyr �IJ������ǻ����������һλ������ϵ���ԭ��֮���SN2��Ӧ���ɻ��ѻ����DZȽϼ��ĺϳɷ�ʽ(Scheme 32)[48].
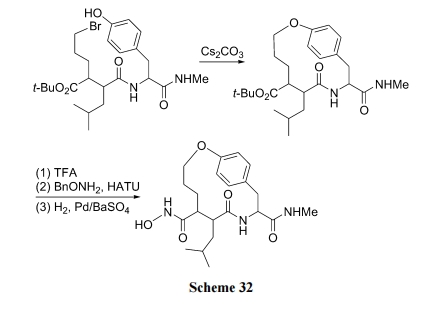
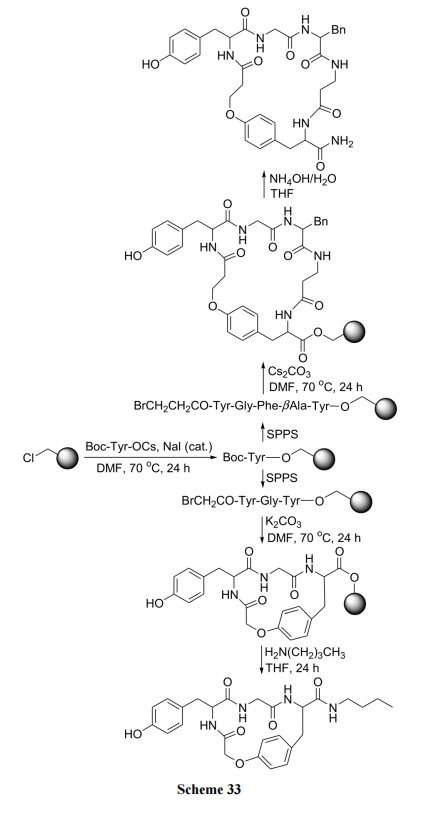
�����ĵ�[49]������ N �˵��������� C �˵� Tyr Ϊ������ͷ, �������ĺϳɷ�ʽ�����Ʊ�����������ͷ�ṹ��ֱ�����м���, ����� K2CO3Ϊ���������ع�����һЩ���ѻ��IJ���(Scheme 33).
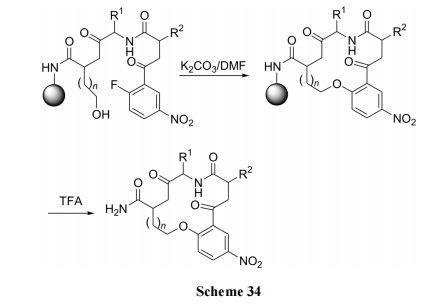
�������ϳɲ�ͬ���ǽ�±ԭ�����ڱ�����, �ǻ�λ��ͬ���ӵ��������. ���ڶ�λ������Ӱ��, F ԭ���нϸߵķ�Ӧ����, �� K2CO3�ļ�������������ɻ��Ѽ�����(Scheme 34)[50].
4.2 ���ѻ��ĺϳ�
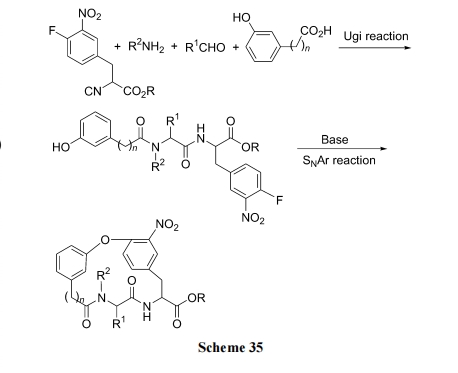
�����Ծ�������������ȡ�������漰������������ֽ��� 4CR (four-component reaction)��ʽ�� Ugi ���Ϸ�Ӧ, �Ƶú����ǻ���������������ֱ�����м���. ����ڼ���·��������� SNAr ��Ӧ, �õ����ѻ��IJ���(Scheme 35)[51].
���������ϳ��������ڡ���λ NO2�� CN ���Ի��±ԭ�ӵĻ�����, ���Լ����ȱ��γ� ��6-��ϩ��������̬�ķ�ʽ��߱�������ԭ�ӵķ�Ӧ����. ����·������ʹ Boc-Phe(4-Cl)OH �����Լ��γɻ����� 36, Ȼ�����Ȼ�������װ�Ľṹ���м��� 37, ������ Na �Լ�����, ��ȥ HCl ���ɷ��Ѽ�. ���ⷴӦ��ȥ��ϵ�����, ��Ŀ�����(Scheme 36)[52].
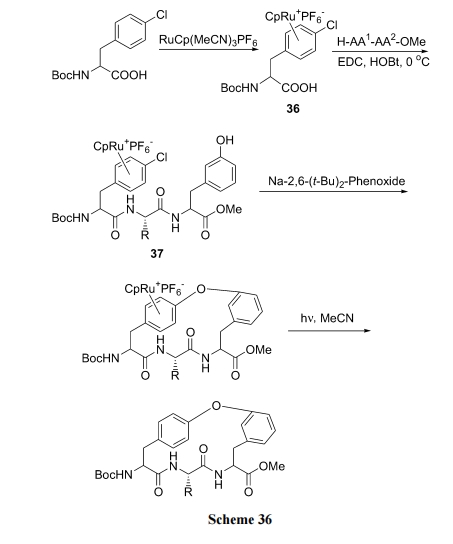
������ Cs2CO3/DMF �����Լ�-�ȱ�����ﷴӦ��ɴ˲�����(Scheme 37)[53].
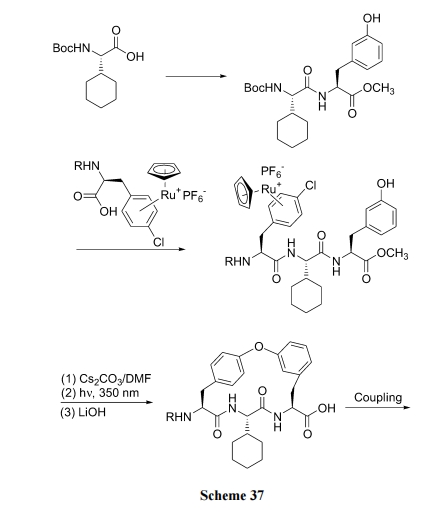
5 �������ѻ��ĺϳ�
����ǰ����ܵľ������������, ���������Žṹ�Ļ��е������š�S�����Ǽ������š�SCH2S�����������š�S��S��S������ʽ. �����Ե����Ž�Ϊ���, �����ַ�Ϊ���������Ѽ��������������. ����������ŵĽṹ��ͬ, �Ʊ������ɷ�Ϊ���¼���: (1)��������Ƭ�ε�������(�� CONH Ϊ��)�Ļ���; (2) SN2 ��Ӧ��ʽ, �������� Cys �л��������ϻ�Ϊ��S����ǰ��ṹ, ��ͬ�����ڵ�±ԭ��Ϊ�˻���; (3) SNAr ��Ӧ��ʽ, ��Cys �������ϻ���ͬ�����ڷ����ϵķ�ԭ��Ϊ���Ϸ�Ӧ����ͷ���.
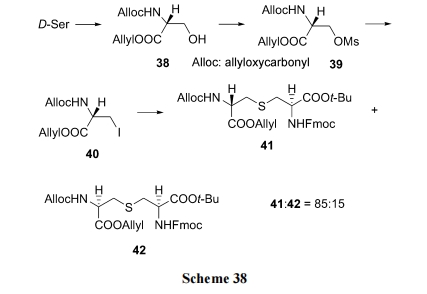
5.1 Ԥ�Ƶ������м���Ļ��ĺϳ�
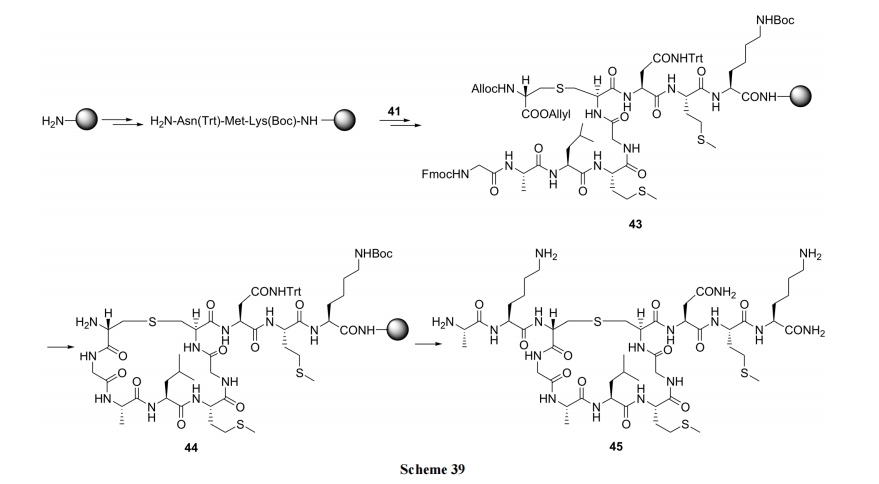
Tabar ��[54]������һ�������Ʊ����������ѡ���Խϸߵĺϳ�·��. �� D-˿����Ϊԭ��, �Ⱥ����ౣ�����ǻ�������������ȡ���õ���Ҫ�м���������������(40). �����ڼ������, ��˫�����İ��װ��ᷢ�� SN2 ��Ӧ, �õ�һ�Բ��ʲ�����Ե��Ե�����Ϊ�ŵ�˫������������ Lanthionine (41). ������Ϊһ���ؼ�������������һ����Ϊ Lantibiotics ����Ȼ�����ĺϳ�(Scheme 38). Tabar ʵ������ 41 ������Ȼ���� Nisin C�������� 45 �Ĺ���ϳ�(Scheme 39)[55].
5.2 �� SN2 ��Ӧ��ʽ�ػ��Ʊ������ѻ���
��SN2��Ӧ��ʽ�ػ����Ʊ������ѻ�����õIJ���. �����ṩ��ԭ�ӵĽṹ��Ϊ�����е� Cys �л�, ����±ԭ�ӵĽṹΪ������������������ N ��������(��������). ����DZ���ķ�Ӧ����, һ����尷[���Ұ���N-�������� DBU (1,8-diazabicyclo[5.4.0]-undec-7- ene)]���Ա�֤ SN2 ��Ӧ�Ľ���. DBU ���Ĺػ��� Eq. 1[56]; ���Ұ�/ˮ���Ĺػ��� Eq. 2[57].
5.3 ������ N ����������������
����������������ЧӦ, ʹ��λ F ԭ�ӻ, ������ SH ���� SNAr ��Ӧ, ���ɷ�������Ѽ�(Scheme 40)[58].
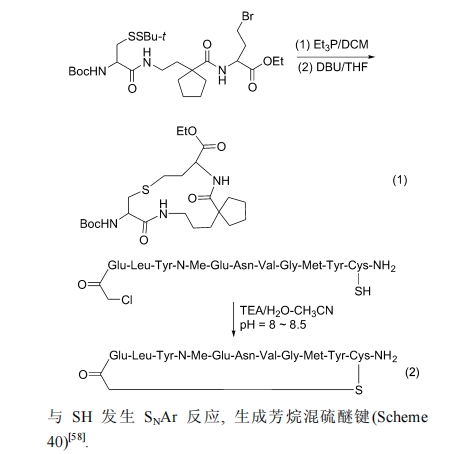
5.4 Michael �ӳɷ�
���������ʵ�λ��� Ser �л��Ͻ���������Ӧ, �������������(Dha), Ȼ���������ϵ����� SH ����Michael �ӳɵõ������ѻ���, ������ѳ������������г�����. ���ַ�ʽ�ֱ���Ϊ��ģ������ϳɵ�һ��;��(Scheme 41)[59].
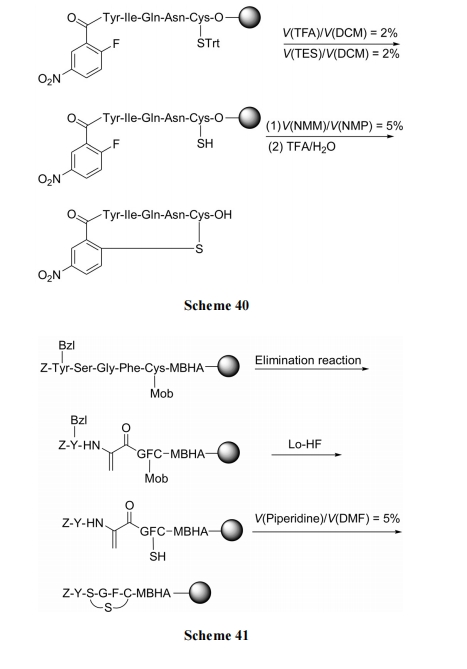
5.5 ���� N �˵�������Զ������ linker ���л����ѽ�
�����ϳɵ��ص������� N �˵�������Զ������linker ���л����ѽⷴӦ. ������е��͵� ��-turn ���Ľṹ(Scheme 42)[60].
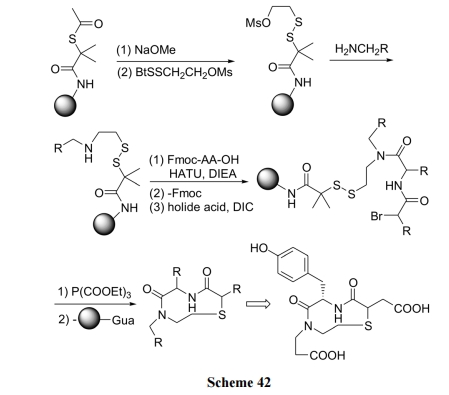
5.6 �����Ǽ�����(SCH2S)��
�����к������ϻ����Ʊ�����������ĵĽṹ����. ���������ת�ƴ����Ķ�������林�����, ���Ի�ܼ� CH2Cl2, ʹ�������ɿ���������������ԭ��֮��, �����Ǽ�����(SCH2S)��(Eq. 3)[61].
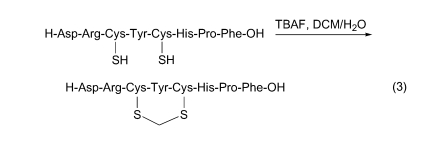
5.7 �������Ż��ĵĺϳ�
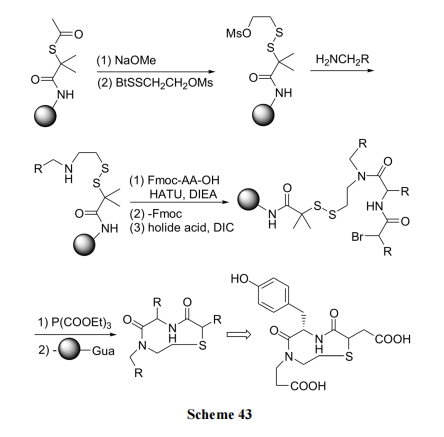
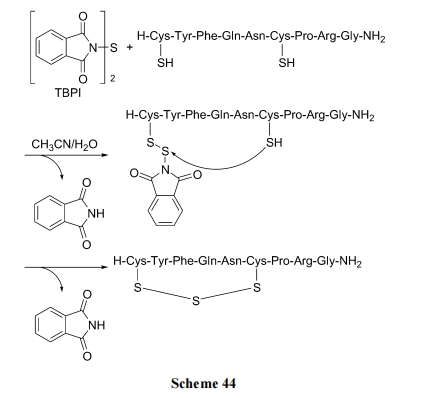
�������Ż��Ĵ���һЩ��Ȼ������, ���Ǿ��еĿ�����ϸ���������Ѿ�����ѧ�ҵĹ�ע. �Ʊ����ֻ��ĵĻ���ԭ�����Ժ������� Cys �л�����Ϊ����ṹ, ���е�һ��Cys�ϵ��ϻ�����ר�ŵ����ѻ��Լ����ɻ������������������. Ȼ���ٽ��ܷ�������һ���ϻ��Ľ���, ��������Ӧ, ���������������. �� TBPI (N,N'-��˫�ڱ��������ǰ�)Ϊ���ѽ������� Scheme 43[62]. �� Mpa(���������������)Ϊ���ѽ�����, �� Scheme 44[63].
5.8 �������ѻ��ĵĺϳ�
���귢��һЩ����5����������������ѵ���Ȼ����, ��˻�ѧ�ҿ�ʼ�Զ������ѻ��ĵĺϳɽ������о�. �Ƚ��д����Ե�һ�ַ��������ȹ��������������װ���л������� 46. Ȼ����һ����Ʒ�ɹ������Լ�BTH ([Bu4N]2S6)��ˮ��Һ�з�Ӧ, ���Է�����ƵôӶ����ŵ������ŵĻ��� 47. ���� BTH ����������Ӧʱ�䲻ͬ, ���Կ������ŵij���(Eq. 4)[64].
6 ��ϩ�����ĺϳ�
��֪�ķ�����, �����ǽ���ϩ��������������, Ȼ���ٽ��з����ڹػ����ֽ�(RCM)��Ӧ������� Heck��Ӧ, ʵ�ֻ���
6.1 ������ RCM ��Ӧ��ʽ
�÷�ʽ���Ⱦ����������齨������������ͷǰ�����˫�����м���. �������л���(Ru)�͵�Grubbs�Լ������·��������� RCM ��Ӧ, ʹ��������.
6.1.1 ˫���� O ԭ����ϩ���ѵ� RCM ����
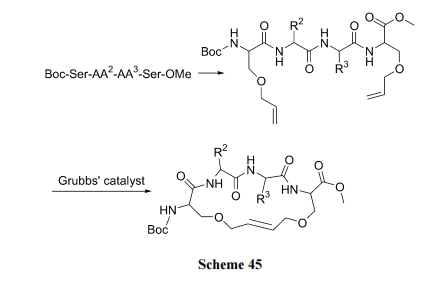
�����ϵ����� Ser, Thr �� Tyr �л��ϵ��ǻ����ʵ���ת����Ӧ(�� SN2, SNAr �� Mitsunobu ��Ӧ)��������ϩ������. Ȼ�� Grubbs �Լ���, ���������� RCM ��Ӧ����ϩ����(Scheme 45)[65].
6.1.2 �����л� ��-̼��ϩ��������� RCM ����
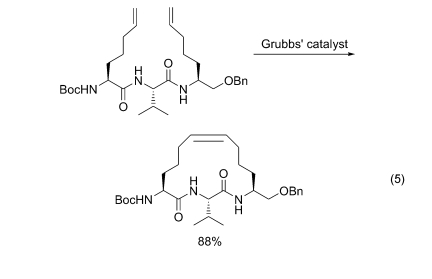
����Ԥ�Ƶĺ�ϩ���������İ��������������Ϊ����, ����������װ�� Grubbs �Լ���, �õ���Ӧ��ϩ����[66](Eq. 5)
6.1.3 ���������� N ԭ����ϩ������� RCM ����
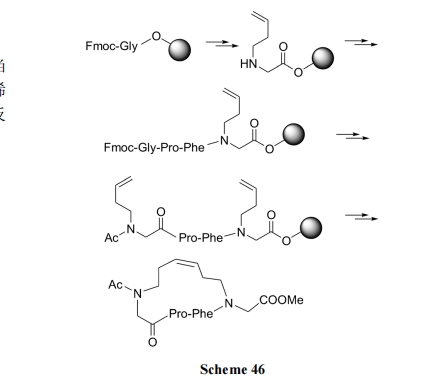
���������� N ԭ����ϩ������� RCM ���ϼ�Scheme 46[67].
6.1.4 �������ϲ��Գ�˫ϩ�� RCM ����
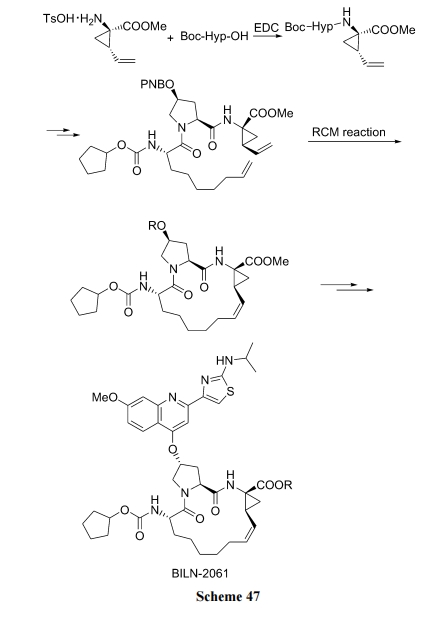
�ѽ����ٴ��� HCV ����ø���Ƽ� BILN-2061 ��һ�����Ļ��ϵĻ�����. �价�ϵ�˫���ž����ڲ�����ϩ���������ϩ֮��� RCM ��Ӧ�γɵ�, �˷�Ӧ���������Ѹߴ� 400 kg ����[68] (Scheme 47).

6.2 �� Heck ��Ӧ�ϳ�ϩ����
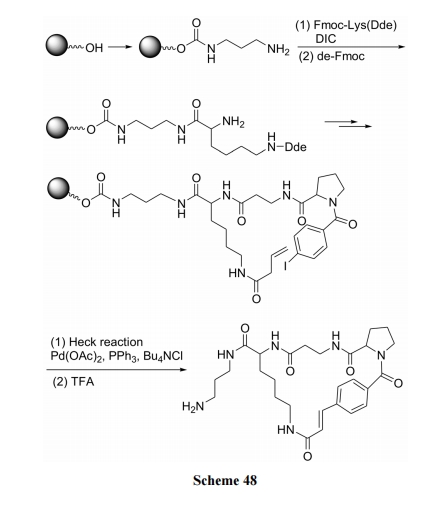
ϩ��Ȳ���ϵIJ�����̼ԭ�ӷ�������ȡ���� Heck��Ӧ���ص�. ����������IJ�ͬλ�÷ֱ�����ϩ(��Ȳ)���������, ��Ϊ�ϻ���ǰ��ṹ. Ȼ�����ٴ���������, ���������� Heck ��Ӧ, ���ɺ�ϩ(��Ȳ)�ŵĻ��IJ���.
ϩ������Լ����ⱽ�ļ��ϼ� Scheme 48[69]; ϩ����������а��ļ��ϼ� Scheme 49[70].

7 �����Ż��ĺϳ�
7.1 ��ȡ��������Ϊ��
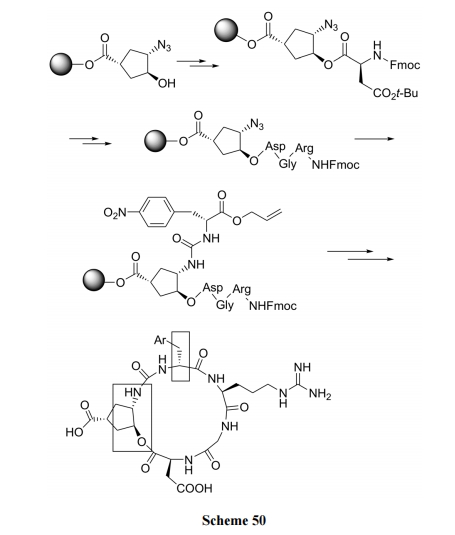
���Ƚ���ȡ��������ͨ���Ȼ����ǻ���֬����, ����Ⱥ��ڻ�Ϊ��λ���ǻ��������Ϸֱ���װ������ N�˶μ� C �˶�. ������ COOH �� NH2���ϳ�Ϊ����������Ի����������ػ���[71] (Scheme 50).
7.2 ����ົ��ṹ�Ļ���
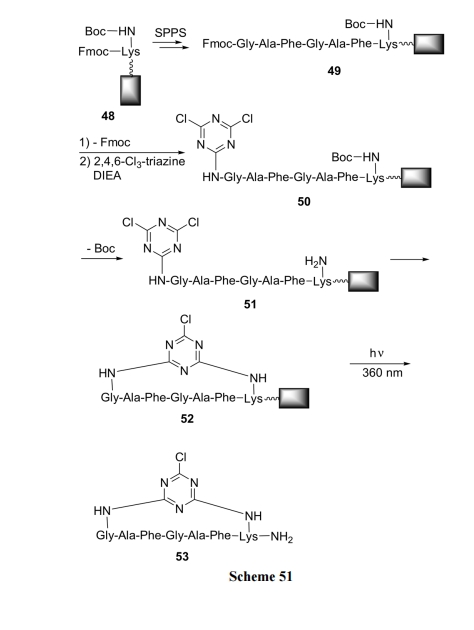
������������ʽ�� Fmoc-Lys(Boc)��Ϊ��װ�����������Ļ���. ���Ƚ� Fmoc-Lys(Boc)ͨ���ʵ��ļ��(Spacer)�ṹ������������ӳ�Ϊ 48. Ȼ���ѳ� Fmoc, ����ֱ����������װ�õ��м��� 49. �ѳ����� N �˵�Fmoc ֮��, ������� NH2 ����������ϵ�һ����ԭ�ӽ��� SN2 ��Ӧ. ��Ȼ������ື����е� Cl �dz�����, �����������ļ�ϡ��ЧӦ, ʹ������ື���ֻ��һ��Cl���ĵ�NH2��Ӧ, �õ�50. ����ѳ�����������ϵ�Boc, ʹNH2����, ��Ϊ51. �ڼ��������, ��ົ��ϵĵڶ��� Cl ԭ������������� NH2���������ڵ� SN2 ��Ӧ, ������ົ�Ƕ��Ļ��� 52[72] (Scheme 51).
7.3 �������˫���ṹ�Ļ���
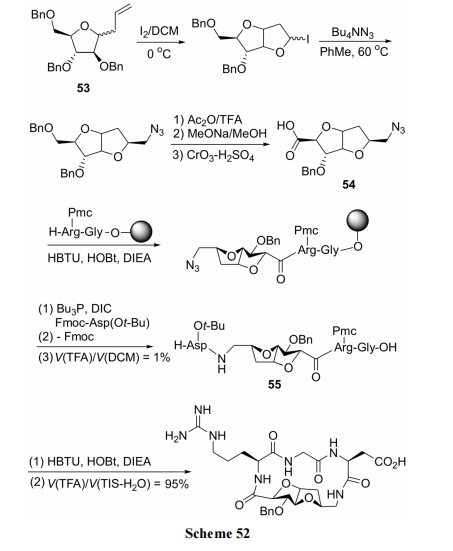
���ȰѸ��Խṹ��˫������������������, �����֧������İ�����л��������������. ����ϳ������������� 53 Ϊԭ��, ������ת����Ӧ�õ����������֧�����м��� 54. ����������֬����. �ٽ���������� N �˵ĵ�����ת��Ϊ NH2, �Ա��� FmocAsp(O-t-Bu)-OH ����, �Ƶû���ǰ��ṹ 55. ����þ����������������ɺ� RGD Ƭ�εĻ����Ʊ�(Scheme 52)[73].
7.4 �Զ��н���Ļ���
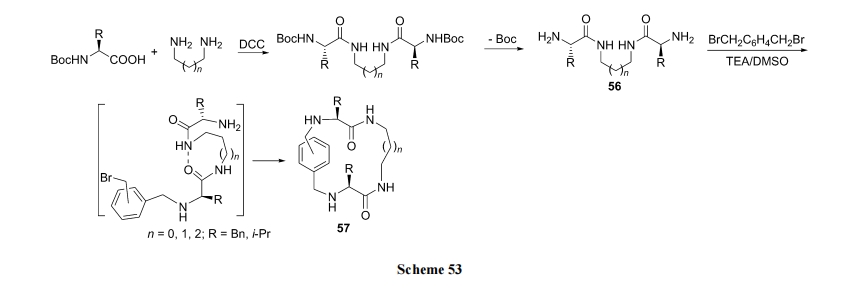
������˫�����ʵ����İ������ ��,��-��������˫������, �����ѳ����˰������� N ������, �õ��ؼ��м���56. Ȼ����ж�λ(���λ)��������56������NH2֮��� SN2 ��Ӧ(������), ���յõ����нṹǶ���˫�ٰ����� 57[74] (Scheme 53).
7.5 ���������������
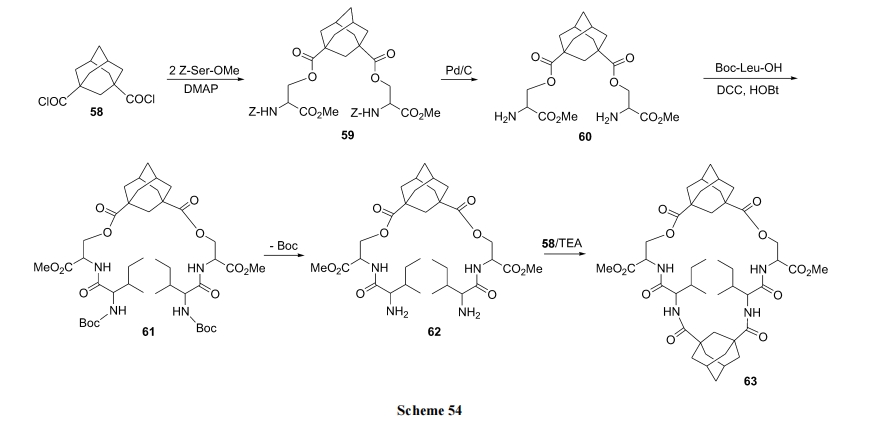
��������������ļ� Scheme 54[75].
8 �١��尷�Ż��ĺϳ�
�١��尷�Ż��ĺϳ���Ҫ���������ַ���: ����Ԥ�ƺ���(��)�����м���, �������������[76,77]; ArF ��H2NAr ֮��ļ��Ϲػ�[78]�Լ�������������� NH2 �ļ��Ϲػ�
9 Freidinger �ֲ�����
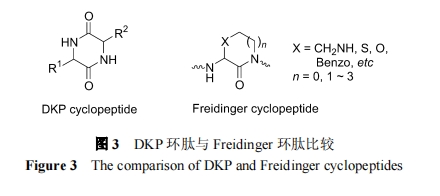
DKP ��ͷ-β�����������������Ļ�����. ���֮��, Freidinger��һ���л��Ħ�-̼ԭ�����Ҳ����ڲл��ĵ�ԭ����������һ����Ļ�����(ͼ 3).���߾����������������˸��Խṹ����, ���ߵĽṹ������ԶԶ���� DKP ����, ��������ڿ����ṹ�ϸ���ҩ(drug like)�Ļ��Ի�����.
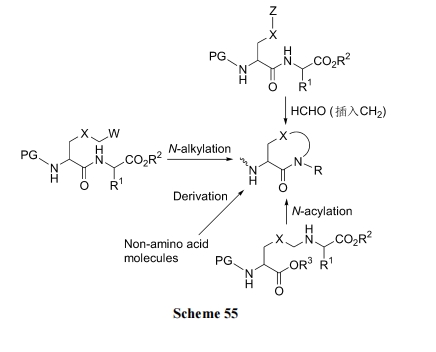
�ϳ� Freidinger ���ĵķ����dz��㷺, ���������Ѿ�ϵͳ�ؽ����˹���[80]. �Ӻ�۲��Խ�, Freidinger ���ĵĺϳɻ����� Scheme 55 ��ʾ 4 ��·��ʵ��.
10 �����ṹ���ĺϳ�
10.1 �����Ż���
����ͨ�л�С��������������ϳ�����, ����ĵĻ��Ϸ���Ҳ�������������(Ϊ��ͷ)֮��� Suzuki ��Ӧ��Ullmann ��Ӧ������ż���ȷ�ӦΪ���[81].
10.2 Mannich ���
������������� Tyr �л�, ����ǻ��� ��-λ������Ϊ������Ĺ����밷��ȩ���� Mannich ����. ʵ�������� N �˰��������ֳɵİ����, ����ʱֻ����ӵ�ȩ���������Ƶ� Mannich ���ͻ���[82].
10.3 PNA ����
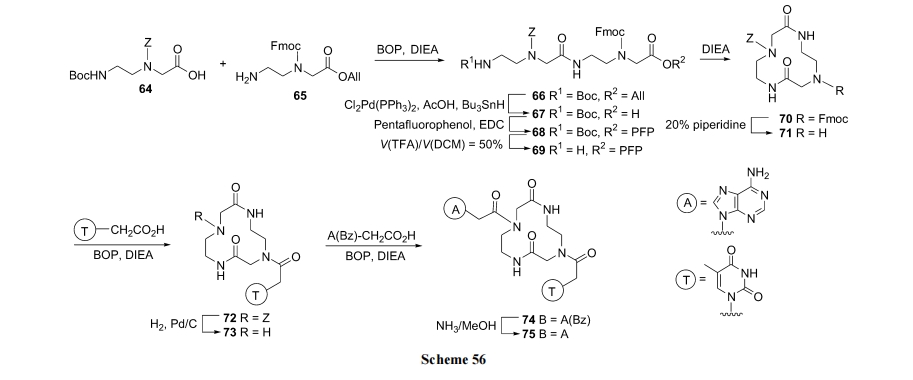
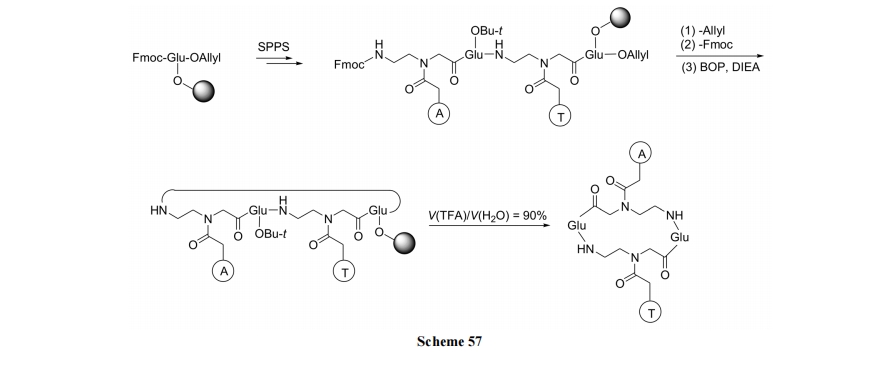
PNA �����һ�-(N-�������)-�ʰ���Ϊ�л������γɵ��ӽ���. �价�����Կɷ�Ϊ���Ʊ�������, �������(Scheme 56)[83]�����Ʊ��������ֱ����, ��(Scheme 57) [84]����.
10.4 ˫ loop ����
������Ȼ�����ļ��˹��ϳɵ��ȵ��������Ժ�����������(loop)Ϊ�ṹ����. ��Щ������������ṹ���в�ͬ, ��ͬһ���������� loop ֮�乲�õ��Žṹ(�ڲ���)������ŵ��Žṹ�����Dz���ͬ��. ���������, �ڲ����ɶ�������ӻ�����; ������������������������ṹ(Scheme 58).
10.3.1 ��Ȼ������ Triostin A ������ĺϳ�
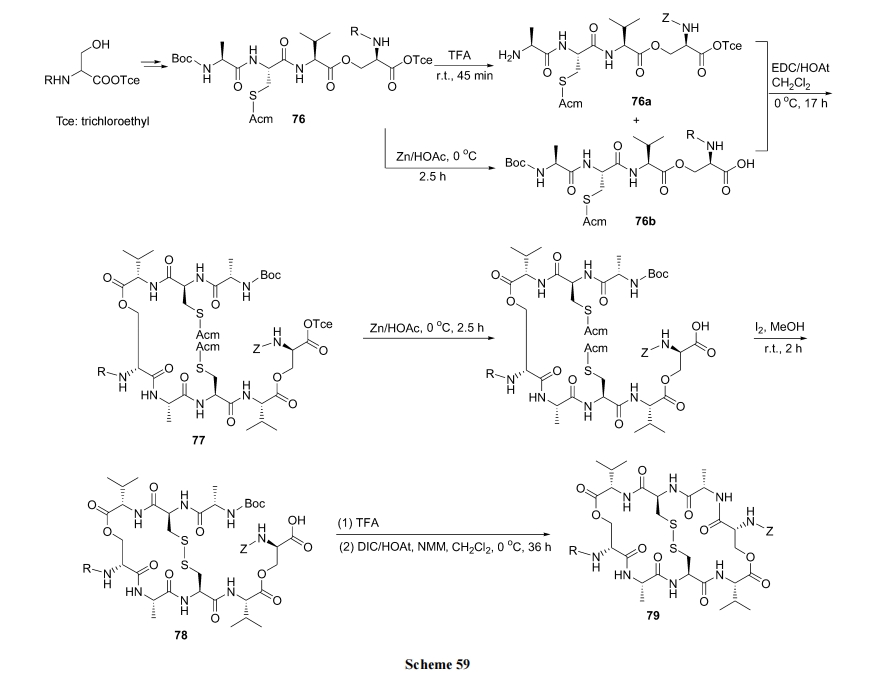
�����Ʊ��� Cys ��ȫ���������� 76. Ȼ��ķ�Ӧ˳�����������ӵ� 76 �������������ϳ�Ϊ 77, �������γɶ����(�ڲ���)�õ��м��� 78. ���һ����ͷ�γ�������(�����), ���˫ loop 79 �Ĺ���(Scheme 59)[85].
10.3.2 ��Ȼ������[Ala7]-Phalloidin �ĺϳ�
���ϳ���Ŀ�껯�����������ỷΪ�ڲ���, ��������Ϊ����ŵ�˫loop����. ��Ҫ�ĺϳɲ��������Ʊ����ڲ��ŵ�Ƭ��, Ȼ���ӳ������. ��������ʹ�Ҳ�CONH �ػ�, ���γ������������, ����˫ loop ����[86].
11 ��
ͳ�Ʊ���, ����ֱ���ĵ�����ԶԶ���ڻ���, ��Ŀǰ���е������ȵ�����������ٴ�����������ѡҩ���Ѿ����е�����ҩ����, ����ȴռһ������. �ɴ˱���, ����ҩ�з���, ���ĵijɹ���ԶԶ����ֱ����. ���, ��ơ��ϳɽṹ�����Ļ��IJ�����һ���ḻ�Ļ�ѧ������, ���ҽ���ٳɸ���������ҩ�ij���.
��������������Ϊ��ҵ����ѧϰ����Ȩ��ԭ����ԭ��־���У�������Ȩ������ϵɾ�������±�ע���������³����������Ķ�ԭ�ļ��ο����ף����Ķ�ԭ��־��
