
еЊвЊ:ИЮдрМВВЁЪЧЭўаВШЫРрЩњУќНЁПЕЕФживЊМВВЁжЎвЛ,ЖдИЮдрМВВЁМьВтМАжЮСЦЗНЗЈЕФбаОПвВв§Ц№ШЫУЧЕФМЋДѓжиЪгЁЃАаЯђвЉЮяЕнЫЭЯЕЭГ(TDDS)ПЩНЋвЉЮябЁдёадЕиЪфЫЭЕНАаЕузщжЏ,ДгЖјЬсИпвЉЮяЕФЩњЮяРћгУТЪВЂНЕЕЭЖОИБзїгУ,вбв§Ц№СЫбаОПепЕФЙуЗКЙизЂЁЃНќФъРД,дНРДдНЖрЕФбаОПГЂЪдНЋАаЯђвЉЮяЕнЫЭЯЕЭГгІгУгкИЮдрМВВЁЕФЯдЯёМьВтвдМАвЉЮя/ЛљвђжЮСЦ,ВЂШЁЕУСЫЪЎЗжЯджјЕФГЩМЈЁЃБОЮФНЋЖдНќФъРДПЊЗЂЕФаТаЭФЩУзИЮАаЯђИјвЉЯЕЭГ,гШЦфЪЧХфЬх-ЪмЬхНщЕМЕФжїЖЏИЮАаЯђИјвЉЯЕЭГдквЉЮя/ЛљвђЕнЫЭвдМАЯдгАМьВтЗНУцЕФзюаТНјеЙзівЛИізлЪі,ЖдИїАаЯђИјвЉЯЕЭГЕФИЮАаЯђФмСІНјааСЫзмНсЖдБШ,ВЂЖдИЮАаЯђИјвЉЯЕЭГЕФЗЂеЙЗНЯђНјааСЫдЄВтЁЃ
ЮвЙњЪЧЪРНчЩЯИЮдрМВВЁЕФИпЗЂЧј,ЦфжаввИЮИаШОепЪ§СПвбОЙ§вк,ЯрЕБгкУП10ИіШЫжаМДга1ИіввИЮИаШОеп,БћИЮВЁЖОИаШОепвВвбГЌЧЇЭђДѓЙи,ЖјвваЭБћаЭВЁЖОЪЧЕМжТИЮгВЛЏЁЂЩѕжСИЮАЉЕФживЊвђЫи[1] ЁЃШЋЙњУПФъГЌЙ§100ЭђШЫЫРгкВЁЖОадИЮбзЕШЯрЙиМВВЁ,ЖјвђИЮАЉЫРЭіЕФШЫЪ§МДГЌЙ§10ЭђШЫ,дкФааджаеМОнАЉжЂЫРЭіШЫЪ§ЕФЕк3ЮЛ,ХЎаджаОгЕк4ЮЛЁЃ
ФПЧА,ЖдгкВЁЖОадИЮбзЁЂИЮгВЛЏЁЂИЮЯЫЮЌЛЏЕШМВВЁШдвдвЉЮяПижЦЮЊжїЁЃЖјЖдгкдчЦкИЮАЉЛМепРДЫЕ(СіПщ аЁгк5cm),ЦфАЉЯИАћВЂЮДРЉЩЂЕНСмАЭНсКЭЩэЬхЕФЦфЫќВПЮЛ,вђДЫЪжЪѕЧаГ§жЮСЦЪЧзюЮЊгааЇЪжЖЮЁЃШЛЖјИЮАЉ ЕФЗЂВЁНЯвўФф,дчЦкЛМепЮоУїЯджЂзД,ЖјИЮАЉЛМепвЛЕЉвђГіЯжжЂзДЖјЧАЭљвНдКОЭея,ЦфВЁГЬДѓЖрвбНјШыжаЁЂЭэЦк,ДэЙ§СЫЪжЪѕжЮСЦЕФзюМбЪБЦк;ДЫЪБМДЪЙЪжЪѕЧаГ§,ЦфжЮСЦГЩЙІТЪвВНіЮЊ20%~30%[2,3] ЁЃгЩДЫПЩМћ,ЪЕ ЯжИЮАЉЕФдчЦкИпСщУєМьВтЪЧЪжЪѕжЮСЦФмЗёГЩЙІЕФЙиМќвђЫи,етЖдгкЯдЯёбЇЕФЗЂеЙЬсГіРДИќИпЕФвЊЧѓ;ЖјЖд гкИЮбзЁЂИЮгВЛЏвдМАжаЭэЦкИЮАЉЛМеп:ЗЧЪжЪѕжЮСЦ,гШЦфЪЧвЉЮяжЮСЦОЭГЩЮЊСЫзлКЯжЮСЦЕФживЊЪжЖЮжЎвЛЁЃ
ЛЏСЦЪЧРћгУЛЏбЇвЉЮяЖдАЉЯИАћВњЩњЩБЩЫзїгУ,ЭЌЪБвВПЩПижЦАЉЯИАћРЉЩЂЁЃФПЧА,ЛЏСЦЪЧжЮСЦИЮАЉЕФзюЦеБщВЩгУЗНЗЈ,ЕЋДЋЭГЕФЛЏСЦЗНЪНЪЧЭЈЙ§ГЃЙцЭООЖИјвЉ,ЕБДяЕНвЛЖЈЕФбЊвЉХЈЖШКѓ,ЛЏСЦвЉЮядкШЋЩэЗжВМ,ШБЗІбЁдёад,дкЩБЩЫАЉЯИАћЕФЭЌЪБвВЖдШЋЩэдрЦїЙЙГЩЩЫКІ,ВЛНігАЯьСЦаЇ,вВИјВЁШЫДјРДОоДѓЭДПр;ЧвЕНДяИЮдржзСіВПЮЛЕФвЉЮяНіЮЊвЛаЁВПЗж,ДѓВПЗжвЉЮяВЛНіЮДЗЂЛгвЉаЇзїгУ,ЛЙЛсЖдЦфЫќе§ГЃдрЦїВњЩњбЯжиЕФЖОИБзїгУ,гАЯьетаЉвЉЮяЕФжЮСЦаЇЙћЁЃвђДЫ,ИФБфЛЏСЦЕФИјвЉЭООЖЁЂБфЛЛвЉЮяМСаЭЕШИїжжИїбљЕФГЂЪдДгЮДМфЖЯ,ЦфжаИЮАаЯђадФЩУзИјвЉЯЕЭГЕФбаОПвбв§Ц№дНРДдНЖрЕФжиЪгЁЃ
ИЮАаЯђвЉЮяЕнЫЭЯЕЭГ(Liver targeted drug delivery system,LTDDS)ПЩНЋвЉЮяЁЂЛљвђЕШЛЏбЇЮяжЪбЁдёадЕиЪфЫЭжСИЮдр,ЬсИпИКдиЮядкИЮдрВПЮЛЕФХЈЖШЁЂбгГЄЦфАыЫЅЦк,ДгЖјДяЕНМѕЩйгУвЉМССПКЭИјвЉДЮЪ§,НЕЕЭвЉЮяЖОИБзїгУ,ЬсИпзЊШОаЇТЪЁЃЫцзХЖдЯИАћБэУцНсЙЙЙІФмШЯЪЖЕФВЛЖЯЩюШы,НќФъРДЪмЬхНщЕМЕФИЮАаЯђЕнЫЭЯЕЭГГЩЮЊбаОПШШЕу[4,5] ЁЃЫќЪЧВЩгУЮяРэЛђЛЏбЇЕФЗНЗЈНЋЬиЖЈЕФХфЛљв§ШывЉЮядиЬх,ЭЈЙ§ХфЛљгыЯИАћФЄЩЯЕФЪмЬхЗЂЩњЬивьЯрЛЅзїгУ,НщЕМЯИАћЪЕЯжЖдаоЪЮгаХфЛљЕФдиЬхВФСЯЕФИпаЇФкЭЬ,ДгЖјДяЕНАаЯђЕнЫЭЕФФПЕФЁЃСэЭт,РћгУLTDDSЛЙПЩНЋЯдгАМСИпХЈЖШЕФИЛМЏгкИЮдрВПЮЛ,ЬсИпЯдгАаЇЙћ,ДгЖјДяЕНЬсИпМьВщСщУєЖШЕФзїгУЁЃгЩДЫПЩМћ,ПЊЗЂИпаЇЕФLTDDSЖдгкИЮдрМВВЁЕФдчЦкМьВтвдМАжЮСЦОљгаЗЧГЃживЊЕФзїгУЁЃЖјФЩУзММЪѕНќ30ФъРДЗЂеЙбИЫй,РћгУФЩУзГпЖШЕФжЌжЪЬхЁЂНКЪјЁЂЮЂФ§НКЕШзїЮЊдиЬхПЩвдЦ№ЕНДйНјвЉЮяШмНтЁЂЬсИпвЉЮяЮШЖЈадЁЂИФЩЦвЉЮяЪЭЗХааЮЊЕШЙІаЇ;ИќживЊЕФЪЧФЩУзММЪѕЛЙПЩвдИФБфвЉЮядкЬхФкЕФЗжВМ,МДОпБИвЛЖЈАаЯђФмСІЁЃвђДЫ,НЋФЩУзММЪѕгыАаЯђЕнЫЭЯЕЭГЯрНсКЯ,ПЊЗЂаТаЭЕФИЮАаЯђадФЩУзИјвЉЯЕЭГГЩЮЊСЫНёФъРДбаОПЕФШШЕуЁЃБОЮФжаНЋЖдФПЧАПЊЗЂЕФжї/БЛЖЏИЮАаЯђИјвЉЯЕЭГНјаазлЪі,ВЂЖдИїАаЯђИјвЉЯЕЭГЕФИЮАаЯђФмСІНјааЖдБШзмНсЁЃ
1 БЛЖЏАаЯђ
БЛЖЏАаЯђ(passive targeted),жИЕФЪЧдивЉЮЂСЃНјШыЬхФкКѓгЩгкжзСігые§ГЃзщжЏМфбЊЙмУмЖШМАЩјЭИадЕФВювьЛђБЛОоЪЩЯИАћзїЮЊЭтНчвьЮяЭЬЪЩЕФздШЛЧуЯђЖјВњЩњЕФЬхФкЗжВМЬиеїЁЃвЛАуШЯЮЊ:БЛЖЏАаЯђЕФЮЂСЃООВТізЂЩфКѓЦфдкЬхФкЕФЗжВМЪзЯШШЁОігкСЃОЖЕФДѓаЁ,аЁгк100nmЕФФЩУзФвЛђФЩУзЧђПЩЛКТ§Л§МЏгкЙЧЫш;100~200nmЕФФЩУзСЃзгПЩИЛМЏгкЪЕЬхжзСіВПЮЛ;0.2~3ІЬmЪБвЛАуБЛИЮЁЂЦЂжаОоЪЩЯИАћЩуШЁ;Дѓгк7ІЬmЕФЮЂСЃЭЈГЃБЛЗЮУЋЯИбЊЙмДВНиСє,НјШыЗЮзщжЏЛђЗЮЦјХн[6] ЁЃгЩДЫПЩМћБЛЖЏИЮАаЯђИјвЉЬхЭГжївЊвРРЕгкЬхФкЕФЕЅКЫЭЬЪЩЯИАћЯЕЭГ(mononuclear phagocytic system,MPS);дчЦкЮФЯзжавВНЋMPSГЦЮЊЭјзДФкЦЄЯЕЭГ(reticulo-endothelial system, RES)ЁЃ
ЕЅКЫЯИАћЗЂЩњгкЙЧЫшЕФЖрФмИЩЯИАћ,бЛЗгкбЊвКжа,ЪЧбЊвКжазюДѓЕФбЊЯИАћЁЃФПЧАШЯЮЊЕЅКЫЯИАћЪЧЭЬЪЩЯИАћЕФЧАЩэ,ЫќДЉЭИбЊЙмФкЦЄНјШызщжЏФк,зЊБфЮЊЭЬЪЩЯИАћЁЃИЮдргЕгаШЫЬхФкЪ§СПзюЖрЕФЭЬЪЩЯИАћЁЊЁЊПнЗёЯИАћ(KCs),дМеМЭЬЪЩЯИАћзмЪ§ЕФ80%ЁЃбаОПБэУїОпгавдЯТЬиеїЕФПХСЃЛсБЛMPSбИЫйЧхç€дкИЮЁЂЦЂЕШЦїЙйРлЛ§,ДгЖјВњЩњвЛЖЈБЛЖЏАаЯђЙІаЇ:(1)гЕгаНЯДѓЕФСЃОЖ(жБОЖ>200nm);(2)ЗЧЧђаЮПХСЃ;(3)БэУцОпгаНЯЧПЪшЫЎад;(4)БэУцОпгаИпУмЖШЕчКЩ[7] ЁЃвђДЫРћгУMPSЭЬЪЩЙІФмОЭГЩЮЊСЫИЮАаЯђИјвЉЕФгааЇЭООЖЁЃChiannilkulchaiЕШНЋИКдиАЂУЙЫиЕФОлЧшЛљБћЯЉЫсвьМКѕЅ(PIHCA)ФЩУзПХСЃ(npc-DXR,СЃОЖ250nm)гУгкаЁЪѓзЊвЦадИЮжзСіЕФжЮСЦ[8,9] ЁЃНсЙћЗЂЯж,ЯрЖдАЂУЙЫизЂЩфвК(f-DXR),npc-DXRПЩУїЯддіМгАЂУЙЫидкИЮдрВПЮЛЕФИЛМЏ,ЦфИЮжзСівЉЪБЧњЯпЯТУцЛ§(AUC0~48h)ЪЧЖдеезщЕФ2.53БЖ;вђДЫЦфвжжЦИЮжзСізЊвЦФмСІдЖдЖИпгкf-DXR(СНепжзСізЊвЦТЪзюДѓЯрВю82%),ДгЖјДѓДѓбгГЄСЫаЁЪѓЕФДцЛюЪБМфЁЃ
ЫфШЛMPSАаЯђЖдгкжЮСЦИЮжзСіОпгавЛЖЈЕФДйНјзїгУ,ЕЋжкЫљжмжЊ,ЖёадИЮжзСіЭљЭљЗЂЩњдкИЮЪЕжЪЯИАћ,ЖјMPSЛсНЋДѓСПвЉЮяОлМЏдкKCsЯИАћжа,етОЭдьГЩДѓСПвЉЮяЮоЗЈжБНгзїгУгкжзСіВПЮЛ,гАЯьжЮСЦаЇЙћЁЃвђДЫ,ПЊЗЂИЮЪЕжЪЯИАћАаЯђИјвЉЯЕЭГОЭЯдЕУгШЮЊживЊЁЃВЛЙ§ЮвУЧвВгІзЂвтЕН:KCsгыИЮВПбзжЂвдМАИЮЯЫЮЌЛЏгазХживЊСЊЯЕ,вђДЫдкжЮСЦДЫРрМВВЁЪБ,MPSЕФБЛЖЏАаЯђзїгУШдШЛОпгаЪЎЗжживЊЕФзїгУЁЃ
2 жїЖЏАаЯђ
жїЖЏАаЯђИјвЉЯЕЭГ(active targeted drug delivery system, aTDDS)ЕФИХФюЪЧгЩЕТЙњПЦбЇМвEhrlichгк1906ФъЪзЯШЬсГіЕФЁЃИУЯЕЭГжївЊАќРЈШ§ИіВПЗж:АаЯђЛљЭХ(targeted ligand)ЁЂдиЬх(carrier)вдМАвЉЮя(drug)ЁЃЦфЛљБОКЌвхОЭЪЧдиЬхНЋвЉЮяЭЈЙ§ОжВПИјвЉЛђШЋЩэбЊвКбЛЗбЁдёадЕФХЈМЏгкАаЦїЙйЁЂАазщжЏЁЂАаЯИАћЛђЯИАћФкНсЙЙЕФИјвЉЯЕЭГЁЃ
ЮЊСЫНЕЕЭMPSЯЕЭГЖджїЖЏАаЯђаЇТЪЕФгАЯь,ЪзЯШгІБЃжЄвЉЮядиЬхФмЬгРыMPSЕФЭЬЪЩ,ВЂЪЕЯждкбЊвКФкЕФГЄаЇбЛЗЁЃвђДЫ,вЛАуОљашЖдФЩУзСЃзгБэУцНјааЧзЫЎИФадВЂПижЦЦфСЃОЖдк100~200nmжЎМф(вргаЮФЯзБЈЕРЮЊ50~150nm)[10] ЁЃгЩгкPEGОпгаИпЧзЫЎадЁЂИпЬхЛ§ХХзшаЇгІвдМАЕЭЖОад,вбОГЩЮЊФПЧАзюГЃгУЕФЧзЫЎИФадЪдМСЁЃ
АДИЮАаЯђЕФАаЯИАћЗжРр , ИЮАаЯђЕнЫЭЯЕЭГПЩЗжЮЊИЮЪЕжЪЯИАћАаЯђ ЁЂЗЧЪЕжЪЯИАћАаЯђвдМАИЮжзСіЯИАћ АаЯђ , ЯТЮФжаНЋЗжБ№ЖдетМИжжИЮАаЯђИјвЉЯЕЭГНјааЯъЯИНщЩм ЁЃ
2.1 ИЮЪЕжЪЯИАћАаЯђ
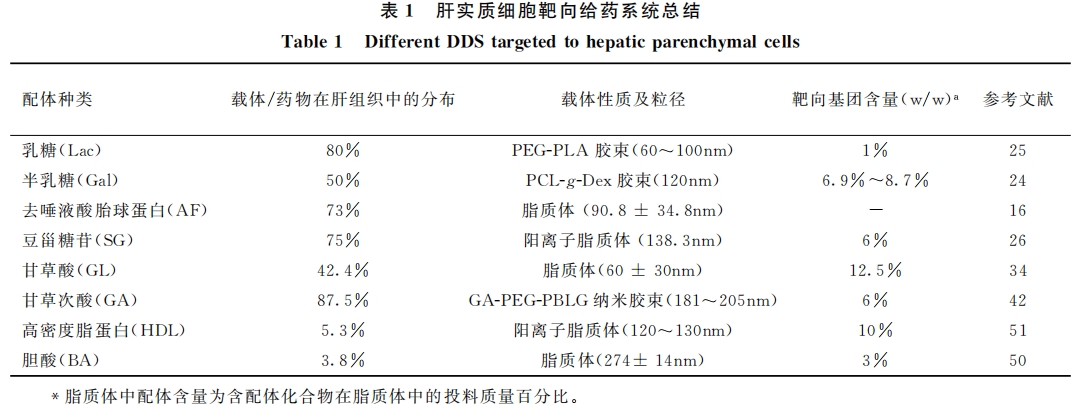
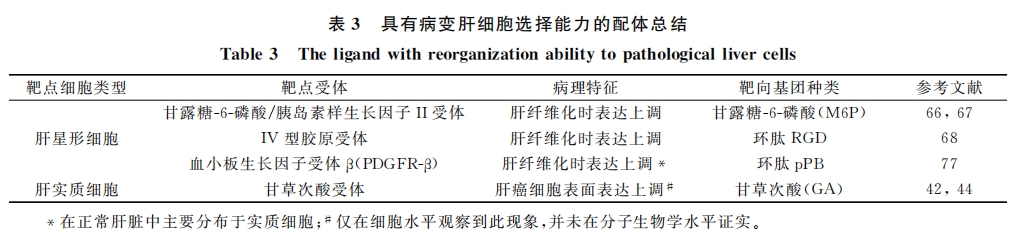
ИЮЪЕжЪЯИАћ(hepatic parenchymal cells, HPCs)ЪЧИЮдржаЪ§СПзюЖрЕФЯИАћ,дМеМИЮдрзмЯИАћЪ§ЕФ60%~70%[11] ЁЃгаЙиИЮЕФДѓЖрЪ§ВЁБфШчИЮАЉЁЂИЮбзЁЂИЮгВЛЏЕШЖрЗЂЩњгкДЫ,ЪЧИЮАаЯђИјвЉЯЕЭГРэЯыЕФАаБъжЎвЛЁЃИЮЪЕжЪЯИАћБэУцКЌгаЖржжЪмЬх,ШчШЅЭйвКЫсЬЧЕААзЪмЬхПЩвдЪЖБ№АыШщЬЧЁЂШщЬЧЕШХфЬх;ИЪВнЫс/ИЪВнДЮЫсЪмЬхПЩвдЪЖБ№ИЪВнЫсвдМАИЪВнДЮЫсЕШЁЃРћгУетаЉЪмЬх-ХфЬхМфЬивьадЕФЯрЛЅзїгУ,баОПШЫдБбаЗЂГівЛЯЕСаИпаЇЕФИЮАаЯђИјвЉЯЕЭГЁЃ
2.1.1 ШЅЭйвКЫсЬЧЕААзЪмЬхНщЕМЕФИЮАаЯђИјвЉЯЕЭГ ФПЧАЪмЬх-ХфЬхНщЕМЕФИЮАаЯђИјвЉЯЕЭГбаОПжа,гІ гУзюЮЊЙуЗКЕФЪЧШЅЭйвКЫсЬЧЕААзЪмЬх(asialoglycoprotein receptor, ASGPR)ЁЃASGPRгжУћИЮФ§МЏЫи (liver lectin)КЭИЮЯИАћАыШщЬЧЪмЬх,ЪЧ20ЪРМЭСљЦпЪЎФъДњгЩAshwellКЭMorell[12] дкбаОПВИШщЖЏЮябЊНЌ ЬЧЕААзДњаЛЪБЗЂЯжЕФвЛжжЪмЬх,ЦфдкВЛЭЌжжЪєЕФВИШщЖЏЮяИЮЯИАћЩЯЕФЪ§СПВЛЭЌ:ШчЪѓИЮЯИАћЩЯКЌга 500,000ИіASGPRЪмЬх[13] ,ШЫИЮЯИАћЩЯКЌга225,000ИіASGPRЪмЬх,ЦфжадМ87%ЗжВМдкИЮЪЕжЪЯИАћФЄ БэУц[14] ЁЃASGPRЪЧЯжНёЮЊжЙСЫНтЕУзюЭИГЙЕФИЮжїЖЏАаЯђЪмЬх,ЫќФмзЈвЛЪЖБ№ФЉЖЫДјгаАыШщЬЧ(Gal)Ва ЛљЛђввѕЃАыШщЬЧАЗ(GalNAc)ВаЛљЕФЙбЬЧЛђЙбЬЧЕААз,ФПЧАвбМћБЈЕМЕФASGPRХфЛљга:ШЅЭйвКЬЧЕААзЁЂШщЬЧЫс(lactoBioni cacid, LA)ЁЂАыШщЬЧЛЏХфЬх(galactosylated ligand,Gal)ЁЂЮоЭйвКЫсЬЅЧђЕААз(AF)вдМА ЖЙчоДМЬЧме(soyBean-derived sterylglucoside,SG)ЕШЁЃ
ЛљгкЩЯЪіЬиад,НќФъРДбаОПепУЧРћгУASGPRХфЛљаоЪЮвЛаЉЭтдДадЙІФмЮяжЪ,ЩшМЦКЯГЩГівЛЯЕСаЛљ гкASGPRЪмЬхНщЕМЕФИЮАаЯђИјвЉЯЕЭГЁЃkimЕШ[15] КЯГЩСЫШщЬЧѕЃЛЏЫЎШмадПЧОлЬЧ,ВЂгУ99mTcНјааБъ МЧ,БъМЧЕФШщЬЧЛЏПЧОлЬЧзЂЩфКѓ10ЁЂ60КЭ120min,УППЫМССПЕФИЮаюЛ§ТЪЗжБ№ЪЧ13.16%ЁЂ16.11%КЭ 16.55%,ИЮАаЯђадУїЯдИпгкЦеЭЈПЧОлЬЧ,ЭЌЪБгжБЛгЮРыАыШщЬЧЫљзшА,БэУїИЮдрЖдЦфЩуШЁЪЧЭЈЙ§ASGPRНщЕМЕФЭООЖЁЃгЩгкДЫЙЄзїПМВьЕФЪЧЫЎШмадИпЗжзгдкИЮдрЕФРлЛ§ГЬЖШЖјЗЧФЩУзПХСЃ,вђДЫПЩвдЛљБОХХГ§ MPSЭЬЪЩЯЕЭГЖдгкЪЕбщНсЙћЕФгАЯь,етЖдгкЦРМлВФСЯЕФжїЖЏИЮАаЯђФмСІЪЧЪЎЗжживЊЕФЁЃ
WuЕШ[16]НЋШЅЭйвКЫсЬЅЧђЕААз(AF)аоЪЮЕНжЌжЪЬхБэУц(AF-L),ВЂбаОПСЫЦфдкаЁЪѓЬхФкЕФЗжВММАЖдИЮЪЕжЪЯИАћЕФАаЯђФмСІ,НсЙћЯдЪО:14CБъМЧЕФAF-LдкИЮдрРлМЦПЩДя73%,ЖјЗЧAFБъМЧЕФжЌжЪЬхдкИЮдрЕФРлМЦНіЮЊ16.5%ЁЃCaiЕШ[17] НЋИЩШХЫи(IFN)гыАыШщЬЧЛЏЕФШЫбЊЧхАзЕААз(HAS)ЙВМлСЌНгЕУЕНХМКЯЮяGal- HSA-IFN,ВЂгУ125IЖдХМКЯЮяНјааБъМЧЁЃНсЙћЯдЪОИЮЯИАћгыGal-HSA-IFN(89.53%)ЕФНсКЯТЪНЯHSA-IFN(6.66%)УїЯдЬсИпЁЃаЁЪѓЬхФкЗжВМбаОПБэУїGal-HSA-IFNОпгаУїЯдЧїИЮад(ИЮдрЗжВМ>45%/g)ЁЃ
SungЕШ[18,19]дкASGPRЪмЬхНщЕМЕФИЮАаЯђИјвЉЯЕЭГСьгђНјааСЫДѓСПбаОП,ВЂШЁЕУСЫЯджјГЩаЇЁЃЫћ УЧЭЈЙ§ГЌЩљШщЛЏЗЈжЦБИСЫБэУцаоЪЮАыШщЬЧАЗЕФОлЙШАБЫс-ОлШщЫс(LA-PGA-PLA)ФЩУзНКЪј(СЃОЖЮЊ 127.5nm),ВЂЬНЬжЦфзїЮЊИЮАаЯђзЯЩМДМдиЬхЕФПЩааад(ШчЭМ1ЫљЪО)[20] ЁЃЫћУЧПМВьСЫФЩУзСЃдкДѓЪѓЬхФк ЕФЗжВМ;ЭЌЪББШНЯВЛЭЌМСаЭзЯЩМДМЖдНгжжHepG2КЩСіТуЪѓЕФжзСіЩњГЄвжжЦФмСІЁЃНсЙћБэУїМДЪЙдкзЂЩф 24hКѓ,ОАыШщЬЧАЗ(LA)аоЪЮЕФФЩУзСЃдкИЮдрЕФИЛМЏТЪШдЯджјИпгкЦфЫќдрЦї,ЧвВЛЫцЪБМфбгГЄЗЂЩњУїЯд БфЛЏЁЃвжСіЪЕбщНсЙћНјвЛВНЯдЪООLAаоЪЮдивЉФЩУзСЃзїгУКѓЕФТуЪѓ,ЦфжзСіЩњГЄЪмЕНУїЯдвжжЦ,ЖјЪѓ ЬхжиМИКѕБЃГжКуЖЈ,ЫЕУїИУдивЉФЩУзСЃФмзЈвЛИпаЇЕиЩБЩЫИЮАЉЯИАћ,ЖјЖде§ГЃзщжЏМИКѕВЛВњЩњгАЯь[21,22] ЁЃ
JeongЕШ[23]ЭЈЙ§вѕРызгПЊЛЗОлКЯЗЈЕУЕНАыШщЬЧаоЪЮЕФОлввЖўДМ-ОлЙШАБЫсмаѕЅЧЖЖЮЙВОлЮя(LA-PEG-PBLG),ЦфПЩдкЫЎШмвКжазщзАГЩ50~300nmЕФНКЪјЁЃзїепЭЈЙ§СїЪНЯИАћвЧБШНЯСЫВЛЭЌжжРрИЮАЉЯИАћЖдгЋЙтБъМЧФЩУзСЃЕФЩуШЁФмСІ,НсЙћЯдЪОHepG2ЯИАћ(ИЛКЌASGPRЪмЬхЕФИЮАЉЯИАћжъ)ФкЕФгЋЙтЧПЖШвЊдЖИпгкSK-Hep01ЯИАћ(ASGPRЪмЬхШБЪЇЕФИЮАЉЯИАћжъ),ЫЕУїASGPRЪмЬхОпгазЈвЛадЁЃ
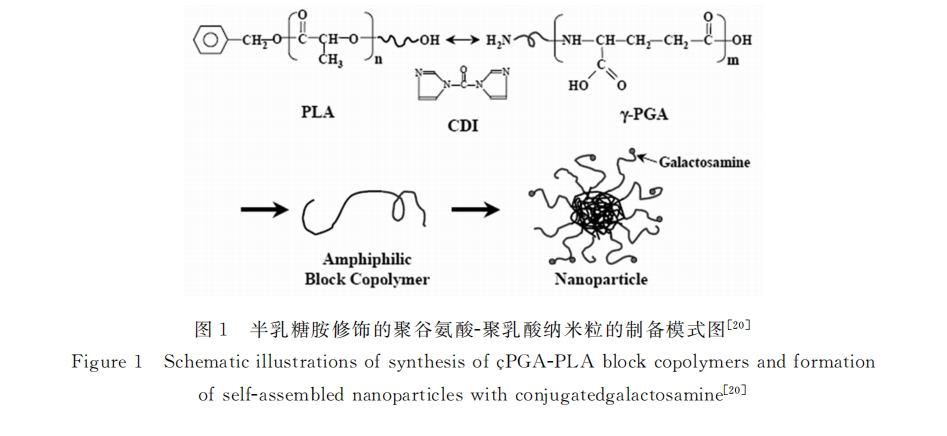
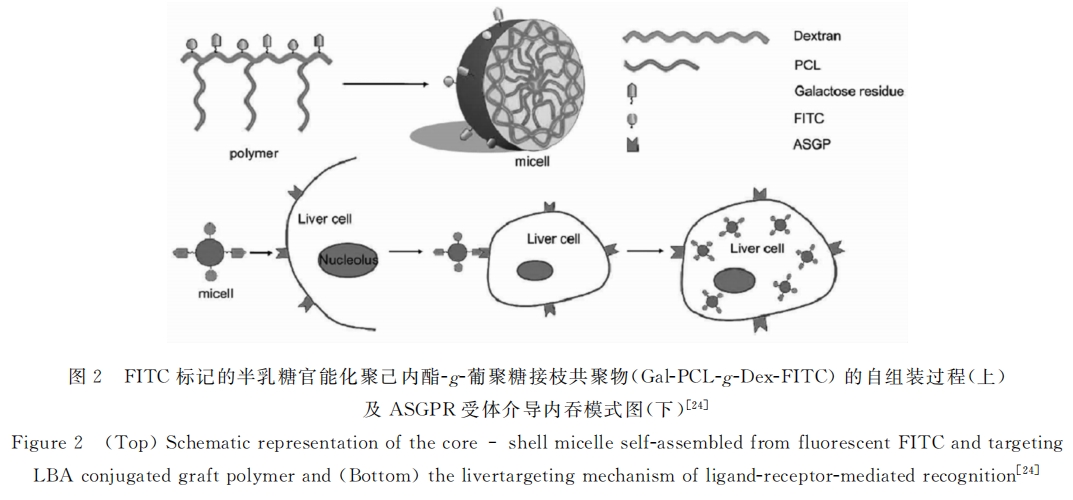
еХЯШе§ПЮЬтзщ[24]жЦБИСЫАыШщЬЧЙйФмЛЏВЂМќНгFITCЕФОлМКФкѕЅ-g-ЦЯОлЬЧНгжІЙВОлЮя(Gal-PCL-g-Dex-FITC),ИУЙВОлЮяВЛТлдкЬхФкЛЙЪЧЬхЭтОљФмаЮГЩЮШЖЈЕФНКЪј(СЃОЖЮЊ120nm)(МћЭМ2)ЁЃНЋАыШщЬЧаоЪЮЕФНКЪјОЮВОВТізЂШыаЁЪѓЬхФк2hКѓ,НЋаЁЪѓНтЦЪ,ИїжївЊдрЦїдШНЌКѓ,ЗЂЯжаЁЪѓИЮдржаЕФТЬЩЋгЋЙтЧПЖШУїЯдИпгкЦфЫќдрЦїЕФгЋЙтЧПЖШ;ЖјЮДаоЪЮАыШщЬЧНКЪјзщаЁЪѓИЮдрдШНЌвКЮДЗЂЯжТЬЩЋгЋЙт,ЫЕУїАыШщЬЧаоЪЮЕФНКЪјОпгаУїЯдЕФИЮдрАаЯђад,НвЪОСЫАыШщЬЧЙйФмЛЏЕФНКЪјдкИЮАаЯђвЉЮяДЋЕнСьгђЕФОоДѓгІгУЧАОАЁЃ
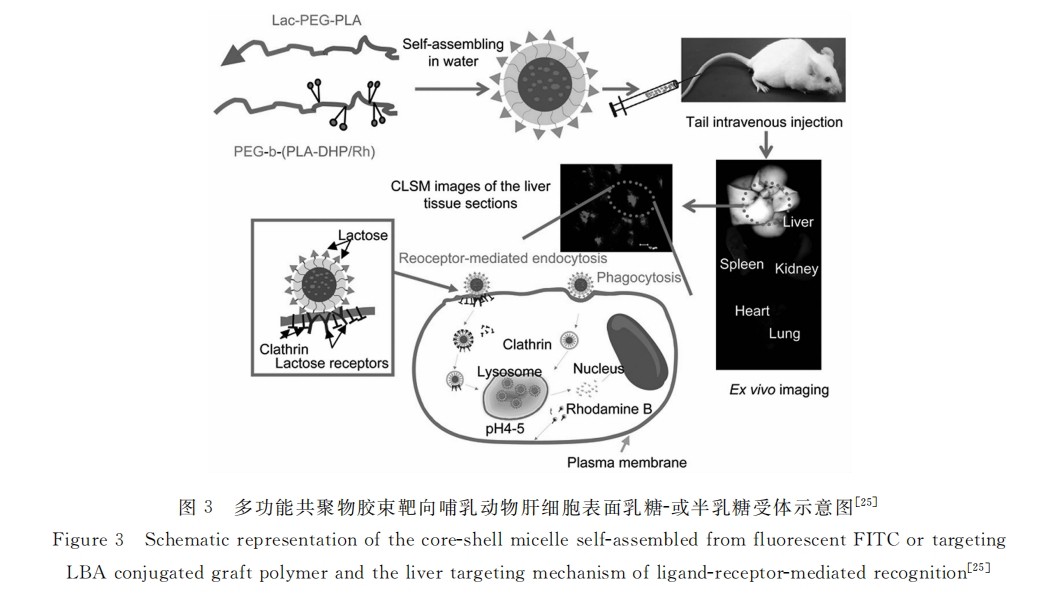
ОАхкБѓПЮЬтзщ[25]жЦБИСЫШщЬЧаоЪЮЕФPEG-PLAЙВОлЮя(Lac-PEG-PLA)КЭТоЕЄУїБъМЧЕФPEG-PLAЙВОлЮя(PEG-b-(PLA-DHP/Rh)),ВЂЭЈЙ§ЙВЛьжЦБИСЫгЋЙтЪОзйЕФНКЪјзїЮЊИЮАаЯђвЉЮядиЬх(60~ 100nm),ЭМ3ЮЊЪЕбщСїГЬЪОвтЭМ,ОЮВОВТіНЋТоЕЄУїБъМЧЕФНКЪјзЂЩфЕНаЁЪѓЬхФк,дкВЛЭЌЪБМфЕуНЋаЁЪѓДІЫР,ШЁГіжївЊдрЦї,ОгЋЙтГЩЯёЗЂЯж,АаЯђНКЪјзщаЁЪѓИЮдрЕФгЋЙтССЖШУїЯдЧПгкЮДаоЪЮШщЬЧзщ,АаЯђзщдк3hЪБИЮдрЕФгЋЙтЧПЖШзюДѓ,дк48hЪБАаЯђзщаЁЪѓИЮдрШдПЩМћЕНКмЧПЕФгЋЙт,ЖјЗЧАаЯђзщвбОЛљБОПДВЛЕНгЋЙтГЩЯё,ЫЕУїШщЬЧаоЪЮЕФНКЪјОпгаУїЯдЕФИЮАаЯђадЁЃ
СэЭт,MaitaniПЮЬтзщ[26]ПЊЗЂСЫвЛРрЛљгкЖЙдДчоДМЬЧме(SG)ЕФИЮАаЯђИјвЉЬхЯЕЁЃЫћУЧНЋSGСДНгЕНжЌжЪЬхБэУц,ВЂНЋЦфгУгкАќдиАЂУЙЫи(DOX)ЁЃЦфбаОПНсЙћЯдЪО,ЯрБШЦеЭЈжЌжЪЬхSGаоЪЮжЌжЪЬхПЩНЋИќЖрЕФвЉЮяЪфЫЭжСИЮЪЕжЪЯИАћ,ЖјSGгыИЮЪЕжЪЯИАћМфЕФНсКЯПЩвдБЛШЅЭйвКЫсЬЅЧђЕААз(AF)вжжЦ,ЬхЯжГіЦфASGPRЪмЬхНщЕМЕФаджЪ[27] ЁЃЪЏОИЕШ[28]вВжЦБИСЫЖЙчоДМЬЧмеаоЪЮбєРызгжЌжЪЬх(SG/PEG-CL),ВЂбаОПСЫЦфдкаЁЪѓЬхФкЕФЗжВММАЖдИЮЪЕжЪЯИАћЕФАаЯђФмСІ,НсЙћЯдЪО:ЮВОВТізЂЩфSG/PEG-CLАыаЁЪБКѓ,ЦфдкИЮдрЕФРлМЦДяЕНЗхжЕ(75%),ЖјЧвЯдЪОГіУїЯдЕФИЮЪЕжЪЯИАћАаЯђФмСІ(ЦфдкЪЕжЪЯИАћжаЕФХЈЖШдМЮЊЗЧЪЕжЪЯИАћжаЕФ2.9БЖ)ЁЃ
2.1.2 ИЪВнЫс/ИЪВнДЮЫсЪмЬхНщЕМЕФАаЯђИјвЉЯЕЭГ ИЪВнЫс(glycyrrhizin acid, GL)КЭИЪВнДЮЫс(glycyrrhetinicacid,GA)ДцдкгкИЪВнЕФИљЁЂОЅВП,АВШЋадИпЁЂСЎМл,ОпгаЖржжвЉРэЛюад,ШчПЙбзЁЂПЙОњЁЂПЙжзСіЁЂПЙРЃбёЁЂПЙИЮВЁЖО/ИЮбзМАБЃИЮЛЄИЮЕШ[29,30] ,НсЙЙШчЭМ4ЫљЪОЁЃЩЯЪРМЭ90ФъДњ,NegishiЕШ[31] ЗЂЯжИЪВнДЮЫсФмгыЪѓИЮдШНЌжаЕФЯИАћФЄзщЗнЮЂСЃЗЂЩњПЩФцадЁЂБЅКЭадНсКЯ,УїШЗжИГіСЫИЮЯИАћФЄЩЯДцдкДѓСПЕФИЪВнДЮЫсЪмЬхЁЃIshidaЕШ[32]дђбаОПСЫЗжРыЪѓИЮЪЕжЪЯИАћЖдИЪВнЫсЕФЩуШЁЛњРэ,жЄЪЕСЫЪѓИЮЯИАћЩЯвВДцдкИЪВнЫсЕФНсКЯЮЛЕуЁЃбюЩНТѓЁЂЙЫдЦцЗаЁзщЪЧЙњФкЪзИіПЊеЙетЗНУцбаОПЕФПЮЬтзщ,ЫћУЧВЩгУНКЬхН№ЬНеыЗЈ,ДгГЌЮЂЫЎЦНдЮЛжЄЪЕСЫДѓЪѓИЮЯИАћФЄЩЯШЗЪЕКЌгаИЪВнДЮЫсКЭИЪВнЫсНсКЯЮЛЕу[33] ,ЫљЕУНсЙћКЭNegishiМА IshidaЕФНсЙћЯрЗћЁЃЛљгкетаЉНсТл,баОПепЭЦВтИЪВнЫс/ИЪВнДЮЫсПЩгУзїИЮАаЯђИјвЉЯЕЭГЕФЕМЯђЛљЭХЁЃгкЪЧ,ЛљгкИЪВнЫс/ИЪВнДЮЫсЪмЬхНщЕМЕФаТаЭИЮАаЯђИјвЉЯЕЭГЕФбаОПж№НЅПЊеЙЦ№РДЁЃ
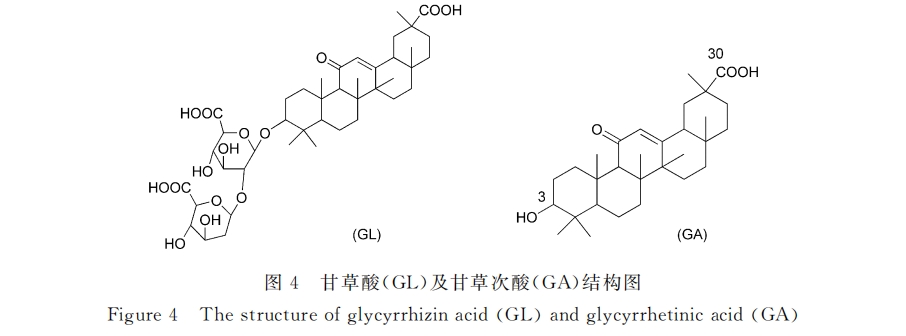
ИЪВнЫсЪмЬх
kiwadaПЮЬтзщ[34,35]ЪзДЮжЦБИСЫИЪВнЫсаоЪЮЕФжЌжЪЬх(GLOS-SUV),ЬхФкЗжВМБэУїИЪВнЫсаоЪЮЕФжЌжЪЬхПЩдкИЮдрИЛМЏ,4hЕФРлЛ§СПЮЊ42.4%,ЪЧЗЧАаЯђзщ(GLOS)ЕФ4БЖ;НјвЛВНбаОПБэУїGLOS-SUVОпгаНЯИпЕФЪѓИЮЯИАћЧзКЭад,2hХрбјКѓ,ЪѓИЮЯИАћЖдGLOS-SUVЕФЩуШЁСПЪЧGLOSЕФ10БЖЁЃЫцКѓ,УЋЩљПЁЁЂКюЪРЯщдкИЪВнЫсаоЪЮЕФИЮАаЯђИјвЉЯЕЭГбаОПжазіСЫЯрЕБЖрЕФЙЄзї[36] ЁЃЫћУЧЯШвдХЃбЊЧхАзЕААзЮЊдиЬх,ИЦЛЦТЬЫиЮЊФЃаЭвЉЮя,жЦБИСЫЮДОИЪВнЫсаоЪЮЕФдивЉФЩУзСЃзг(Cal-BSA-NP)КЭИЪВнЫсаоЪЮЕФдивЉФЩУзСЃ(Cal-BSA-NP-GL),НсЙћБэУї2hКѓЪѓИЮЯИАћЖдCal-BSA-NP-GLЕФЩуШЁСПЪЧCal-BSA-NPзщЕФ4.43БЖЁЃЦНЦфФмЁЂСжАЎЛЊЕШ[37,38]дђвдПЧОлЬЧЮЊдиЬх,ЭЈЙ§РызгНЛСЊЗЈжЦБИСЫБэУцаоЪЮИЪВнЫсЕФПЧОлЬЧФЩУзСЃ(CS-NPs-CL),ЬхЭтЯИАћЪЕбщНсЙћБэУї:CS-NPs-CLПЩЬивьадЕидкИЮЪЕжЪЯИАћИЛМЏ,ИЛМЏТЪЮЊИЮЗЧЪЕжЪЯИАћЕФ4.9БЖ,ЧвГЪЯжУїЯдЕФЪБМфКЭМССПЫЋживРРЕад,ЫЕУїИЪВнЫсЪмЬхНщЕМЕФФкЭЬЖдИЮЪЕжЪЯИАћОпгаИпЖШЬивьадЁЃетвЛНсТлОпгаКмживЊЕФвтвх,вђЮЊДѓЖрЪ§ВЁБфШчИЮАЉЁЂИЮбзЁЂИЮгВЛЏЖрЗЂЩњгкИЮЪЕжЪЯИАћ,вђДЫбажЦФмЙЛЬивьАаЯђИЮЪЕжЪЯИАћЕФИјвЉЯЕЭГИќОпЪЕМЪвтвхЁЃ
ИЪВнДЮЫсЪмЬх
дкNegishiбаОПжаЗЂЯж[31] :ИЪВнДЮЫсгыИЮЯИАћЕФНсКЯФмСІдЖдЖДѓгкИЪВнЫс,вђДЫ,вдИЪВнДЮЫсзїЮЊИЮАаЯђХфЬхгаЭћЛёЕУИќЮЊгХвьЕФАаЯђЙІФмЁЃУЋЩљПЁЁЂКюЪРЯъ[39]зюдчбаОПСЫДЫРрИЮАаЯђдиЬх,ЫћУЧвдЛьКЯРржЌ(СзжЌЁЂЕЈЙЬДМ)ЮЊдиЬх,жЦБИСЫИЪВнДЮЫсаоЪЮЕФдиИЦЛЦТЬЫижЌжЪЬх(Cal-LP-GA),ВЂбаОПСЫЦфЖдЪѓдДЯИАћЕФЧзКЭФмСІЁЃДѓЪѓИЮЯИАћФкЭЬЪЕбщЯдЪО:ЪѓИЮЯИАћЖдCal-LP-GAЕФЩуШЁСПЪЧЦеЭЈжЌжЪЬхЕФ3.3БЖ,ВЂЧвгЮРыИЪВнДЮЫсФмЯджјЕФвжжЦИУЙ§ГЬ,ЖјЖдеезщМИКѕВЛЪмгАЯь,жЄЪЕСЫИЪВнДЮЫсЪмЬхНщЕМЕФИЮАаЯђФмСІЁЃ
ЩЯЪібаОП,дкЪѓдДЯИАћЫЎЦНжЄЪЕСЫИЪВнДЮЫсНщЕМЕФИјвЉЬхЯЕОпгаУїЯдЕФИЮАаЯђФмСІ,ЕЋФПЧАЮЊжЙИЪВнЫс/ИЪВнДЮЫсНщЕМЕФАаЯђИјвЉЯЕЭГМИКѕШЋЖМЭЃСєдкЪЕбщЪвНзЖЮ,КмЖрЮЪЬтгаД§НјвЛВНЕФЬжТл,Шч:(1)GL/GAЪмЬхНщЕМЕФАаЯђИјвЉЬхЯЕЪЧЗёЖдШЫдДИЮЯИАћОпгаЭЌбљЕФЧзКЭФмСІ;(2)АаЯђдиЬхНЋвЉЮяДѓСПОлМЏдкИЮдрВПЮЛЕФЭЌЪБ,ЪЧЗёЛсЖде§ГЃИЮзщжЏЫ№ЩЫ;(3)ИЮдрМВВЁЛМепЭљЭљАщЫцзХДњаЛФмСІЯТНЕЕШЮЪЬт,ЬхФкЛЗОГЛсЗЂЩњНЯДѓИФБфЁЃвђДЫдкдЮЛИЮжзСіЬѕМўЯТ,ИУАаЯђИјвЉЬхЯЕФмЗёе§ГЃЗЂЛгвЉаЇвВЪЧашвЊНјвЛВНЬНЬжЕФЮЪЬтЁЃЛљгкетаЉЮЪЬт,БОПЮЬтзщЩшМЦСЫвЛЯЕСаИЪВнДЮЫсЪмЬхНщЕМЕФИЮАаЯђИјвЉЯЕЭГ,ВЂЖдЦфИЮАаЯђФмСІЁЂЬхФкЭтвжСіаЇЙћЁЂвЉДњЖЏСІбЇМАЦфЖджзСізщжЏМАе§ГЃзщжЏЕФЫ№ЪЇЧщПіНјааСЫГЄЦкЁЂЯЕЭГЕФбаОПЁЃВщШ№ЬЮЕШ[40,41]ГЩЙІжЦБИСЫвЛЯЕСаЖЫЛљХМСЊИЪВнДЮЫсЕФОлЙШАБЫсмаѕЅИЮАаЯђВФСЯвдМАВрСДХМСЊИЪВнДЮЫсЕФПЧОлЬЧФЩУзСЃзг,ВЂЖдЦфжЦБИЬѕМўЁЂЮШЖЈадМАЪЭвЉааЮЊНјааСЫЯЕЭГбаОПЁЃдкДЫЛљДЁЩЯЛЦЮЂЕШ[42]РћгУПЊЛЗОлКЯжЦБИСЫИЪВнДЮЫс-ОлввЖўДМ-ОлЙШАБЫсмаѕЅЕФдиАЂУЙЫиНКЪј(DOX/GA-PEG-PBLG)ВЂПМВьСЫЦфЖдШЫИЮАЉ7703ЯИАћЕФЧзКЭФмСІ,НсЙћЯдЪОИЪВнДЮЫсЖдШЫдДИЮЯИАћЭЌбљОпгаКмЧПЕФЧзКЭад,7703ЯИАћЖдАаЯђзщНКЪјЕФЩуШЁФмСІЪЧЗЧАаЯђзщЕФ3.7БЖ(МћЭМ5)ЁЃЭЌЪБЯИАћЖОадЪЕбщБэУїТувЉАЂУЙЫи(DOX)ЁЂЗЧАазщ(DOX/PEG-PBLG)КЭАаЯђзщ(DOX/GA-PEG-PBLG)ЕФIC50жЕЗжБ№ЮЊ90ЁЂ82КЭ47ng/mLЁЃИЪВнДЮЫсаоЪЮЕФдивЉНКЪјЕФIC50НЯТувЉDOXНЕЕЭСЫдМвЛАы,втЮЖзХDOX/GA-PEG-PBLGФмдкДяЕНКЭТувЉЯрЭЌСЦаЇЕФЧщПіЯТПЩМѕЩйвЉЮягУСП,ДгЖјНЕЕЭвЉЮяЖОИБзїгУ,ЬсИпВЁШЫвРДгЖШЁЃДЫЭт,ЫЮЯрШнПЮЬтзщ[43] жЦБИСЫИЪВнДЮЫсаоЪЮИКдиЛљвђжЌжЪЬх,НсЙћБэУїИЪВнДЮЫсаоЪЮКѓПЩЯджјЬсИпжЌжЪЬхЖдШЫИЮАЉЯИАћHepG2ЕФзЊШОаЇТЪ,ЭЌбљжЄЪЕСЫИЪВнДЮЫсЖдШЫдДИЮЯИАћЕФАаЯђФмСІЁЃ
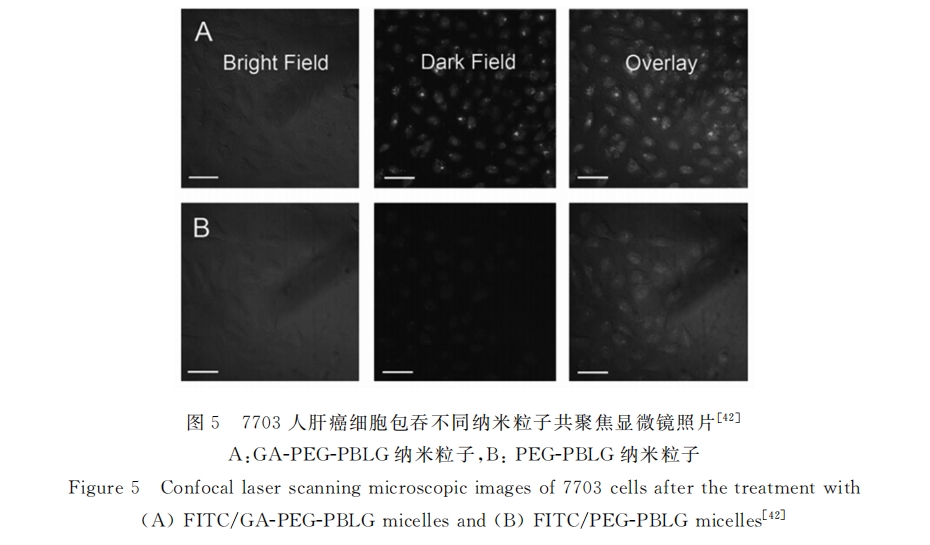
дкжЄЪЕСЫИЪВнДЮЫсЖдШЫдДЯИАћЕФАаЯђФмСІКѓ,БОзщдкЖЏЮяЬхФкМЬајбаОПСЫИЪВнДЮЫсвЉЮядиЬхЕФЧїИЮФмСІМАДњаЛЧщПіЁЃЬяЧиЕШ[44]РћгУ99mTcБъМЧСЫИЪВнДЮЫс-ОлввЖўДМ/ПЧОлЬЧ(GA-PEG/CTS)ИДКЯФЩУзСЃ,ВЂдкДѓЪѓЬхФкПМВьСЫПеАзФЩУзСЃзгЕФЗжВМааЮЊ,НсЙћБэУї, GA-PEG/CTSИДКЯФЩУзСЃПЩИпЖШИЛМЏдкИЮдр,еМзмМССПЕФ51.3%,ЪЧЖдеезщ(ЮДаоЪЮGAЕФОлввЖўДМ/ПЧОлЬЧИДКЯФЩУзСЃ)ЕФ2.6БЖЁЃЮтГЌЁЂЙљЮАгЂ[45]вВЕУЕНСЫРрЫЦНсЙћ:гыШЅАпђњЫиШмвКзщЯрБШ,ЦфжЦБИЕФИЪВнДЮЫсаоЪЮЕФШЅАпђњЫижЌжЪЬх(Gal-GAOStNC-LP)дкИЮдрЕФАаЯђжИЪ§ЮЊ5.2,ОпгаУїЯдЕФИЮдрАаЯђад,ФмЬсИпвЉЮяСЦаЇ,НЕЕЭЖОИБзїгУЁЃЫцКѓ,ЬяЧиЕШ[46]гжРћгУЕЅЙтзгЗЂЩфМЦЫуЛњЖЯВуГЩЯёЪѕММЪѕ(SPECT)ПМВьСЫGAаоЪЮФЩУзСЃЕФИЮАаЯђЛњРэ,ЖдGAЕФНсКЯЮЛЕуНјааСЫЗжЮі,НсЙћжЄЪЕДгВЛЭЌЮЛЕу(3ЮЛєЧЛљМА30ЮЛєШЛљ,МћЭМ4)СЌНгЕФGAаоЪЮФЩУзСЃЖдДѓЪѓИЮдрОљОпгаКмЧПЕФИЮАаЯђадЁЃ
ЮЊСЫбаОПИЪВнДЮЫсЪмЬхНщЕМИЮАаЯђИјвЉЬхЯЕЖдвЉЮяДњаЛЕФгАЯьЁЃеХДГФъЕШ[47]жЦБИСЫИЪВнДЮЫсаоЪЮКЃдхЫсФЦдиАЂУЙЫиФЩУзСЃзг(DOX/GA-ALGNPs),ВЂПМВьСЫИЮАаЯђИјвЉЬхЯЕЕФвЉДњЖЏСІбЇЁЃбаОПНсЙћЯдЪО:ЮВОВТізЂЩф3hКѓ(7mg/kg)аЁЪѓИЮдрвЉЮяХЈЖШДяЕНЗхжЕ(67.8ІЬg/g),ЪЧЗЧАаЯђФЩУзСЃзгзщЗхжЕвЉЮяХЈЖШЕФ2.8БЖ;ДЫЭт,ЯрЖдАЂУЙЫибЮЫсбЮ(DOXЁёHCl)зЂЩфвК,АаЯђзщвЉЮядкИЮдрЕФЦНОљзЄСєЪБМф(MRT)МААыЫЅЦкЯджјЩЯЩ§,ЗжБ№ЪЧDOXЁёHClЕФ9.7БЖКЭ3.8БЖ,МДЪЙдкзЂЩфвЉЮя4ЬьКѓ,ИЮдржаШдПЩМьВтЕННЯИпХЈЖШЕФАЂУЙЫи(26.7ІЬg/g)ЁЃгЩДЫПЩжЊDOX/GA-ALGNPsВЛНіПЩвдДйНјвЉЮядкИЮдрЕФИЛМЏ,ЖјЧвбгГЄСЫвЉЮядкИЮдрВПЮЛЕФзЄСєЪБМф,ЯджјЬсИпСЫвЉЮяЕФЩњЮяРћгУТЪЁЃ
БОзщЧАЦкбаОПжаЗЂЯж:АќдиАЂУЙЫиЕФGA-PEG/CTSИДКЯФЩУзСЃФмЯджјвжжЦвьЮЛИЮАЉКЩСіТуЪѓЕФжзСіЩњГЄЁЃЕЋжкЫљжмжЊ,ИЮдрЪЧШЫЬхживЊЕФДњаЛЦїЙй,ЦфЗЂЩњВЁБфКѓ,ШЫЬхЛЗОГЛсЗЂЩњКмДѓИФБфЁЃвђДЫ,дкдЮЛИЮжзСіФЃаЭЬѕМўЯТбаОПИЮАаЯђИјвЉЬхЯЕЕФаЇЙћОЭЯдЕУгШЮЊживЊЁЃеХДГФъЕШЪзЯШНЈСЂСЫдЮЛИЮжзСіКЩСіаЁЪѓФЃаЭ,ВЂдкДЫЛљДЁЩЯбаОПСЫDOX/GA-ALGNPsЕФЖддЮЛжзСіЕФвжжЦаЇЙћ[47] ЁЃНсЙћЯдЪО:DOX/GA-ALGNPsЖдгкдЮЛИЮжзСівжСіТЪПЩДя76.6%(wt),ЖјЯрЭЌИјвЉЬѕМўЯТЕФDOXЁёHClвжСіТЪЮЊ52.6%(wt);ЭЌЪБ,ЮвУЧЗЂЯжзЂЩфDOXЁёHClзщаЁЪѓаФМЁЯИАћгаЛЕЫРЯжЯѓЧвЬхжигаЯджјЯТНЕ,ЖјАаЯђзщаЁЪѓаФМЁЯИАће§ГЃЧвЬхжиЮоУїЯдБфЛЏЁЃНсКЯвдЩЯНсЙћПЩжЊ,гЩгкDOX/GA-ALGNPsПЩНЋвЉЮяМЏжаЪфЫЭЕНИЮдрВПЮЛВЂМѕЩйЦфдкЦфЫќдрЦїЕФЗжВМ(ШчаФдр),вђДЫЦфВЛНіПЩЯджјЬсИпЦфИЮжзСівжжЦФмСІ,ЖјЧвУїЯдНЕЕЭвЉЮяЕФЖОИБзїгУЁЃ
дкбаОПжаЮвУЧЗЂЯжСЫвЛИігаШЄЕФЯжЯѓ:ЫфШЛDOX/GA-ALGNPsНЋвЉЮяМЏжаЪфЫЭЕНСЫИЮдр,ШЛЖјзщжЏЧаЦЌНсЙћЯдЪОЦфЖде§ГЃИЮЯИАћВЂЮоУїЯдЩЫКІ[47] ЁЃМјгкДЫБОзщеЙПЊСЫНјвЛВНбаОПЁЃЬяЧиЕШ[48]баОПСЫИЪВнДЮЫсаоЪЮПЧОлЬЧСђЫсѕЅНКЪј(DOX/GA-SCTS)гыШЫИЮАЉЯИАћ(HepG2)вдМАе§ГЃШЫИЮЯИАћ(ChangLiver)ЧзКЭадЕФВювьЁЃЪЕбщНсЙћЯдЪО:HepG2ЯИАћЖдDOX/GA-SCTSЕФАќЭЬФмСІЪЧChangLiverЯИАћЕФ2.18БЖ;ЖјЗЧИЮЯИАћ(HelaЯИАћ)DOX/GA-SCTSФЩУзСЃзгвдМАЗЧИЪВнДЮЫсаоЪЮФЩУзНКЪј(DOX/SA-SCTS)ЕФАќЭЬЮоЯджјВюБ№(ШчЭМ6ЫљЪО)ЁЃетБэУїИЪВнДЮЫсЪмЬхВЛНіОпгаИЮЯИАћЧзКЭФмСІ,ЖјЧвЛЙОпгаЪЖБ№ИЮАЉЯИАћКЭе§ГЃИЮЯИАћЕФЙІФм,вђДЫИЪВнДЮЫсЪмЬхНщЕМЕФвЉЮяЕнЫЭЯЕЭГПЩНЋвЉЮяжївЊдЫЫЭЕНИЮжзСіВПЮЛ,ДгЖјМѕЩйЖде§ГЃИЮзщжЏЕФгАЯь,етЖдгкЬсИпвЉЮяРћгУТЪЁЂНЕЕЭвЉЮяЖОИБзїгУОпгаЪЎЗжживЊЕФвтвхЁЃ
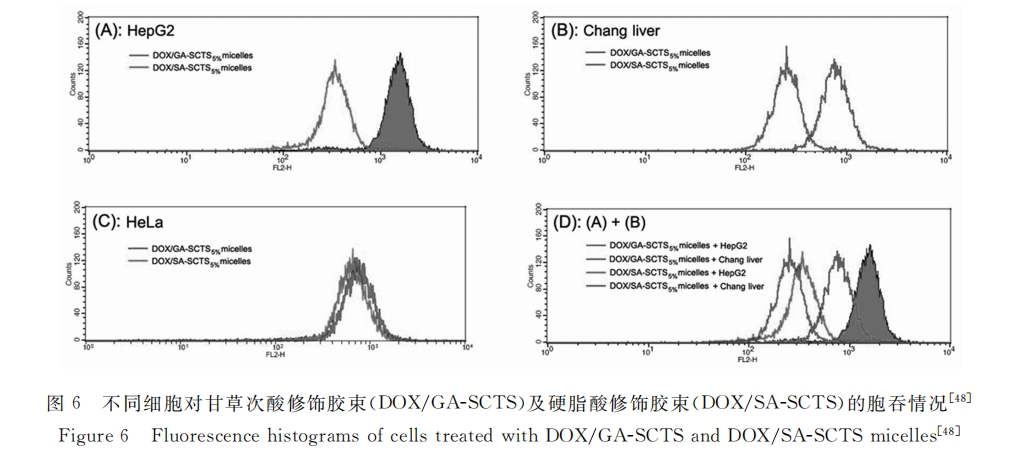
злЩЯЫљЪі,етаЉНсЙћВЛНіНјвЛВНВћУїСЫИЮЯИАћБэУцДцдкИЪВнРрЮяжЪЕФЪмЬх,вВжЄЪЕСЫИЪВнЫс/ИЪВнДЮЫсЪмЬхНщЕМЕФИЮАаЯђИјвЉЕФПЩФмадЁЃгыШЅЭйвКЫсЬЧЕААзЪмЬхвЛбљ,ИЪВнЫс/ИЪВнДЮЫсНщЕМЕФАаЯђИјвЉЯЕЭГжївЊгУгкИЮЪЕжЪЯИАћИјвЉ,ЖјЧв,ИЪВнЫс/ИЪВнДЮЫсНщЕМЕФАаЯђИјвЉЯЕЭГПЩдкдЮЛИЮжзСіФЃаЭжаЗЂЛггХвьЕФжЮСЦаЇЙћ,ОпгаЪЎЗжСМКУЕФгІгУЧАОАЁЃЕЋИУАаЯђИјвЉЯЕЭГЛЙгааэЖрД§НтОіЕФЮЪЬт,ЬиБ№ЪЧИЪВнЫс/ИЪВнДЮЫсЕФЪмЬхШдЮДШЗЖЈ,ЧвХфЬх-ЪмЬхМфЯрЛЅзїгУЛњРэвВЛЙашНјвЛВНВћУїЁЃ
2.1.3 ЕЈЫс(бЮ)НщЕМЕФИЮАаЯђИјвЉЯЕЭГ ЕЈЫс(BileAcid)ЪЧЕЈжЕФжївЊГЩЗж,дкЬхФкОпгаЬиЪтЕФзЊдЫЯЕЭГ,ПЩБЛИЮдрЬивьадЮќЪе,етжжЮќЪеЪЧЭЈЙ§ИЮЯИАћФЄЩЯЕФNaЪЎвРРЕадзЊдЫЯЕЭГ(NTCP)МАNaЪЎЗЧвРРЕадзЊдЫЯЕЭГ(OATP)РДЪЕЯжЕФ[49] ЁЃвдЕЈЫсЮЊвЉЮядиЬх,ВЛЕЋФмЙЛЪЕЯжвЉЮяЕФИЮАаЯђад,МѕЩйЖОИБзїгУ,ЛЙФмЙЛЬсИпвЉЮяЕФПкЗўЩњЮяРћгУЖШЁЃ
ГТжОХєЕШ[50]жЦБИСЫЕЈЫсаоЪЮжЌжЪЬх(BP2BL)ВЂбаОПСЫЦфИЮАаЯђФмСІ,НсЙћЯдЪОгыДЋЭГжЌжЪЬх(CL) ЯрБШBP2BLЕФбЊвКбЛЗЪБМфМАИЮдрзЄСєЪБМфОљЯджјбгГЄ,ЦфдкИЮдрЕФвЉЪБЧњЯпЯТУцЛ§жЕ(AUC)ЪЧCLЕФ1.45БЖ,ЬхЯжГівЛЖЈЕФИЮАаЯђФмСІЁЃЕЋбаОПЯдЪО,ИУИјвЉЬхЯЕВЂЮДЬсИпвЉЮядкИЮдрЕФЗхжЕХЈЖШЁЃ
2.1.4 ЧхЕРЗђЪмЬхНщЕМЕФАаЯђИјвЉЯЕЭГ ЧхЕРЗђЪмЬх(Scavenger receptor,SR)ЪЧЙЬгаУтвпжавЛРрживЊЕФФЃЪНЪЖБ№ЪмЬх,ЗжЮЊЖржжРраЭ(SRAЁЂSRB),ЦфжаSRBжївЊЗжВМгкИЮдрВПЮЛ,ВЮгыжЌЗОЁЂЕЈЙЬДМзЊдЫЕШЬхФкЙ§ГЬЁЃИпУмЖШжЌЕААз(HDL)ЕФжївЊЙІФмЪЧЧхГ§бЊвККЭЯИАћжаЙ§ЖрЕФЕЈЙЬДМ,ЫќПЩНЋГСЛ§дкбЊЙмБкЕФЕЈЙЬДМЁЂбЊаЁАхПХСЃАўРыЯТРД,ШЛКѓЭЈЙ§гыИЮЯИАћБэУцSRBЕФИпЧзКЭадНсКЯНЋЕЈЙЬДМДјШыИЮдр,ОЕЈЕР-ГІЕРХХГіЬхЭт,вђДЫHDLОпгавЛЖЈЕФИЮЯИАћАаЯђЙІФмЁЃ
гЩгкжЌЕААзЬсДПВНжшИДдгЁЂдьМлМЋИп,КмФбдкЪЕМЪЩњВњжагІгУ,вђДЫбаОПепЖрВЩгУЛљвђжизщММЪѕЛёЕУ жЌЕААзВЂгІгУгкЪЕбщЪвбаОПЁЃLeeЕШ[51]РћгУЛљвђММЪѕжЦБИСЫжизщШЫАЂЦгжЌЕААзA1(rhapoA-1ЁЂapoA-1ЪЧHDLЕФжївЊРраЭ),ВЂвдrhapoA-1аоЪЮбєРызгжЌжЪЬхЮЊдиЬхИКдиsiRNAгУгкБћаЭИЮбзЕФжЮСЦ(СЃОЖ 120~150nm)ЁЃЬхФкЪЕбщНсЙћЯдЪО:rhapoA-1ИЮАаЯђФмСІТдИпгкбЊНЌжаЕФЬьШЛapoA-1,ЖјСНепаоЪЮжЌ жЪЬхОљПЩЯджјдкИЮдрИЛМЏЁЃRuiЕШ[52] НЋЯдгАМС(Gd)-DTPAСЌНгдкжизщИпУмЖШжЌЕААзПХСЃБэУц(Gd- chol-HDL),ПМВьЦфдкИЮКЫДХ(MRI)ЯдЯёжаЕФаЇЙћЁЃНсЙћЯдЪО,Gd-chol-HDLПЩБЛHepG2ЯИАћАћЭЬ,Цф жаЬхФкПЩбИЫйИЛМЏгкИЮдрМАЪЎЖўжИГІВПЮЛ,ЯдЪОСЫЦфзїЮЊИЮЛђЪЎЖўжИГІMRIЯдЯёЕФЧАОАЁЃ
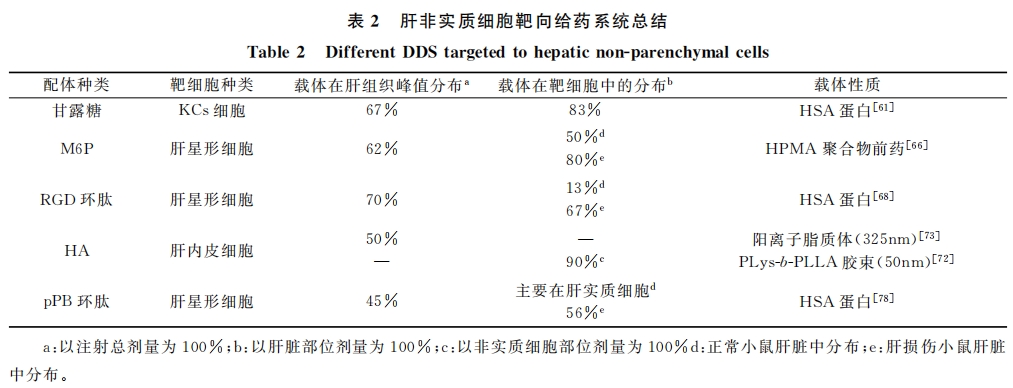
Бэ1жаЖдАаЯђИЮЪЕжЪЯИАћЕФИјвЉЯЕЭГНјааСЫзмНс,ДгБэжаПЩМћASGPRИЮАаЯђИјвЉЯЕЭГгЕгаКмИпЕФИЮАаЯђФмСІ,ЦфНщЕМИјвЉЬхЯЕдкИЮдрЕФИЛМЏТЪДяЕНСЫ50%~80%ЁЃШЛЖј,габаОПБэУїASGPRЕФУмЖШКЭНсКЯЛюадЛсЫцаэЖрЩњРэКЭВЁРэЬѕМўЕФБфЛЏЖјЗЂЩњИФБф,ВЂЧвДѓЖрЪ§ИЮдрМВВЁЛМепЕФбЊЧхжаОљДцдкНсКЯвжжЦМС,ПЩФмЛсЕМжТASGPRНсКЯЛюадЕФНЕЕЭ,ЪЙЦфЖдАыШщЬЧХфЛљЕФЬивьадЪЖБ№зїгУМѕШѕ[53,54] ,вђДЫЦфдкВЁБфФЃаЭЬхФкЕФИЮАаЯђФмСІЛЙгаД§НјвЛВНЕФЪЕбщбщжЄЁЃИЪВнЫс/ИЪВнДЮЫсаоЪЮЕФИЮАаЯђИјвЉЯЕЭГвВОпгаЗЧГЃЧПЕФЧїИЮад,ЦфгыASGPRЬхЯЕЕФИЮАаЯђФмСІЯрЕБЛђТдгагХЪЦ(42%~87%)ЁЃСэЭтбаОПБэУї:GAНщЕМЕФИЮАаЯђЬхЯЕОпгаИЮАЉЯИАћЬивьбЁдёад,етЖдгкПЊЗЂаТаЭИЮАЉеяЖЯЯдгАМСМАНЕЕЭвЉЮяЖде§ГЃИЮзщжЏЕФЩЫКІЖМОпгаЪЎЗжЛ§МЋЕФзїгУЁЃ
СэЭт,ШЫУЧдјЖдЕЈЫсМАЧхЕРЗђЪмЬхНщЕМЕФИЮАаЯђИјвЉЯЕЭГМФгшКёЭћ,вВКмЖрЮФЯзБэУїЫќУЧгыИЮЪЕжЪЯИАћгаНЯЧПЕФНсКЯ,ЕЋЦфдкЬхФкВЂУЛгаЬхЯжГігХвьЕФИЮАаЯђФмСІ(ИЮИЛМЏТЪ<10%)[55] ЁЃдьГЩетжжЧщПіЕФжївЊдвђЪЧ:ЫфШЛЕЈЫсМАHDLжївЊВЮгыИЮдрВПЮЛЕФЮяжЪзЊдЫ,ШЛЖјЮоЗЈдкИЮдрГЄЪБМфзЄСє,ДгЖјЫќУЧгАЯьСЫвЉЮядкИЮдрЕФЛ§Рл,вђДЫвВгабаОПепНЋШщЬЧ/АыШщЬЧЕШаоЪЮдкЦфБэУцНјааИФадвдЬсИпЦфЧїИЮад[56,57]ЁЃ
2.2 ИЮЗЧЪЕжЪЯИАћАаЯђ :
ИЮдржаГ§СЫЪЕжЪЯИАћЭтЛЙга30%зѓгвЕФЗЧЪЕжЪЯИАћ,ЫќУЧдкИЮдржавВЦ№зХживЊЕФзїгУЁЃИЮдржаЕФЗЧЪЕжЪЯИАћжївЊАќРЈПнЗёЯИАћ(Kupffer cells, KCs)ЁЂИЮёМФкЦЄЯИАћ(sinusoidal endothelial cells, SECs)вдМАИЮаЧзДЯИАћ(hepatic stellate cells, HSCs)ЕШЁЃЦфжаKCsЪЧОпгаЭЬЪЩФмСІЕФОоЪЩЯИАћ,дМеМИЮЯИАћзмСПЕФ15%,ЪЧЭЬЪЩЯЕЭГжаЪ§СПзюЖрЕФЯИАћ(дМеМ80%);баОПБэУїKCsгыИЮВПбзжЂЗЂЩњвдМАИЮЯЫЮЌЛЏОљгаживЊСЊЯЕЁЃЖјHSCsдкИЮдрЪмЕНбзжЂЛђЛњаЕДЬМЄЕШЫ№ЩЫЪБЛсБЛМЄЛю,ВЂЭЈЙ§діЩњКЭЗжУкЯИАћЭтЛљжЪВЮгыИЮЯЫЮЌЛЏЕФаЮГЩКЭИЮФкНсЙЙЕФжиНЈ[58] ЁЃSECsдђдкМИжжМБТ§адИЮВЁЕФЗЂВЁЛњРэжаОљЦ№ЕНживЊзїгУ,ФПЧАжївЊзїЮЊПЙИЮбзвЉЮяЕФАаБъ[59] ЁЃвђДЫ,ЖдИЮЗЧЪЕжЪЯИАћАаЯђИјвЉЯЕЭГЕФбаОПЖдгкжЮСЦИЮбзЁЂИЮЯЫЮЌЛЏМАИЮгВЛЏЕШМБ/Т§адМВВЁгазХживЊЕФвтвхЁЃБОЮФНЋжївЊЖдНќЮхФъРДЗЧЪЕжЪЯИАћИЮАаЯђвЉЮяЕнЫЭЯЕЭГЕФзюаТНјеЙНјаазлЪіВЂЖдЦфИЮАаЯђадФмНјааЖдБШЁЃ
2.2.1 ИЪТЖЬЧЪмЬхНщЕМЕФИЮАаЯђИјвЉЯЕЭГ ИЪТЖЬЧЪмЬх(mannose receptor, MR)ЪЧЗжзгСПЮЊ175,000ЕФПчФЄЕААз,ФмгыКЌгаИЪТЖЬЧХфЛљЕФЮяжЪЬивьадЪЖБ№НсКЯ,ЙуЗКДцдкгкИЮKCs ЯИАћБэУц[60,61] ЁЃBijsterBoshЕШ[62]НЋИЪТЖЬЧаоЪЮЕФЦЯЬбФдмеѕЅУИОЪѓЮВОВТізЂЩф,10minКѓМьВтЕНВФСЯдкИЮдрЕФИЛМЏТЪ ЮЊ65.6%ЁЃ
HirataЕШ[61]жЦБИСЫВЛЭЌГЬЖШИЪТЖЬЧЛЏЕФШЫбЊЧхАзЕААзЭЛБфЬх(Man-rHSAs: D63N, A320T and D494N),вдМАЫћУЧЕФШ§БЖЬх(TM-rHSA: D63N/A320T/D494N),ВЂЭЈЙ§111InБъМЧЗЈМьВтСЫЦфдкЖЏЮяЬхФкЕФЗжВМЧщПіЁЃНсЙћЯдЪО:ИїHASЭЛБфЬхОљПЩбИЫйДгбЊвКжаЧхç€РлМЦЕНИЮдр,ЦфЧхГ§ЫйТЪгыИЪТЖЬЧЛЏГЬЖШГЩе§ЯрЙи,ЖјЧвНќ90%ЕФTM-rHSAБЛИЮЗЧЪЕжЪЯИАћАћЭЬЁЃНгЯТРДзїепгжПМВьСЫTM-rHSAдкВЛЭЌЗЧЪЕжЪЯИАћ(KCsЯИАћМАSECsЯИАћ)жаЕФЗжВМ,НсЙћБэУї125IБъМЧЕФTM-rHSAМИКѕЭъШЋгыKCsЯИАћНсКЯ,ЖјКмЩйгыSECsЯИАћНсКЯ,БэУїИЪТЖЬЧЪмЬхжївЊЗжВМгкKCsЯИАћБэУцЁЃ
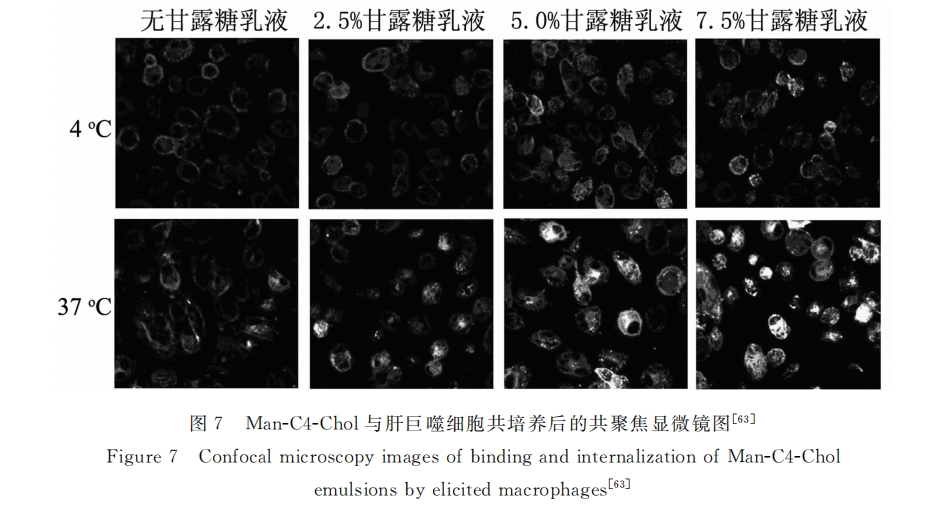
НЋИЪТЖЬЧдиЬхгІгУгкЛљвђжЮСЦСьгђ,KawakamiМАHashidaПЮЬтзщзіСЫДѓСПЙЄзїЁЃyeeprae[63] ЕШВЩгУO/WШщЛЏЗЈжЦБИСЫИЪТЖЬЧаоЪЮАќКЌДѓЖЙгЭЁЂEggPCКЭЕЈчоЯЉЕФШщвК(Man-C4-Chol),ПМВьСЫВЛЭЌКЌСПИЪТЖЬЧЕФMan-C4-CholЖдИЮОоЪЩЯИАћЕФАаЯђаЇЙћЁЃЭМ7ЮЊТоЕЄУїБъМЧMan-C4-CholгыИЮОоЪЩЯИАћЙВХрбјКѓЕФЙВОлНЙЯдЮЂОЕНсЙћ,баОПЗЂЯж,ИЪТЖЬЧУмЖШдНДѓ,Man-C4-CholЖдИЮОоЪЩЯИАћ(KCs)ЕФАаЯђаЇЙћдНКУ,ЧвИЪТЖЬЧЪмЬхНщЕМЕФАаЯђОпгаЮТЖШвРРЕадЁЃжЎКѓ,ИУзщгждкИУЬхЯЕжав§ШыPEG2000,жЦБИСЫИЪТЖЬЧаоЪЮЕФбєРызгХнзДжЌжЪЬх(Man-PEG2000BL,дМ 150nm)[64] ЁЃНсЙћЯдЪО:ЯрЖдPEG2000-BL,Man-PEG2000-BLПЩУїЯдЬсИпЛљвђЖдИЪТЖЬЧЪмЬхБэДяЯИАћЕФзЊШыаЇТЪ,ЖјЦфдкЖЏЮяИЮдрЕФРлЛ§ГЬЖШвВУїЯдИпгкЦфЫќЦїЙй(ПЩДя67%)ЁЃНќРД,ИУзщгжвдИУдиЬхЮЊЛљДЁ,баЗЂСЫАќдиЯИАћФк№ЄИНвђзг1(ICAM-1)ИЩШХаЁRNA(siRNA)ЕФMan-PEG2000BL,ВЂНЋЦфгУгкМБадИЮбзЕФжЮСЦ[65] ЁЃ
2.2.2 ИЪТЖЬЧ-6-СзЫс/вШЕКЫибљЩњГЄвђзгЪмЬхНщЕМЕФИЮАаЯђИјвЉЯЕЭГ е§ГЃЧщПіЯТИЮаЧзДЯИАћ(HSC)ДІгкОВжЙзДЬЌ,ЕБИЮдрЪмЕНбзжЂЛђЛњаЕДЬМЄЕШЫ№ЩЫЪБ,ИЮаЧзДЯИАћБЛМЄЛюВЂзЊЛЏЮЊГЩЯЫЮЌЯИАћЯИАћ,вђДЫHSCМЄЛюЪЧЕМжТИЮЯЫЮЌЛЏЕФживЊвђЫиЁЃHSCМЄЛюКѓ,ЦфБэУцИЪТЖЬЧ-6-СзЫс/вШЕКЫибљЩњГЄвђзгЪмЬх(mannose-6-phosphate/insulin-likegrowth factor II receptors,M6P/IGFII receptor)БэДяЩЯЕїЁЃРћгУетжжЯжЯѓ,баОПепНЋM6PжБНгСЌНгЕНВФСЯБэУц,ИГгшЦфИЮАаЯђЙІФмЁЃ
ШчYangЕШ[66] вдpHУєИаЖрыФGFLGЮЊСЌНгБлНЋM6PМќСЌЕНОлєЧБћЛљМзЛљБћЯЉѕЃАЗ(PHEMA) ЩЯ,ВЂИКдиIаЭНКдЬивьШ§БЖЬхЙбКЫмеЫс(M6P-GFLG-HPMA-GFLG-32P-TFO)гУгкИЮЯЫЮЌЛЏЕФжЮСЦЁЃНсЙћЯдЪО:M6P-GFLG-HPMA-GFLG-32P-TFOПЩбИЫйБЛИЮдрЮќЪе,ЖјЦфжага80%ЪЧБЛИЮаЧаЮЯИАћАќ ЭЬ,БэУїM6P/IGFIIЪмЬхНщЕМдкДЫЙ§ГЬжаЦ№ЕНСЫживЊзїгУЁЃPrakashЕШ[67]НЋЖрМлM6PМАПЙАЉвЉЮяАЂУЙ ЫиЭЌЪБаоЪЮЕНШЫбЊЧхАзЕААзЩЯ(Dox-HSA-M6P28)ВЂНЋЦфгУгкжзСіжЮСЦЁЃЬхФкЗжВМНсЙћЯдЪО,гаНќ50% ЕФDox-HSA-M6PОлМЏдкИЮдрЁЃ
2.2.3 IV аЭНКдЪмЬхНщЕМЕФАаЯђИјвЉЯЕЭГ ИЮЯЫЮЌЛЏЛМепЕФИЮаЧаЮЯИАћБэУц,VIаЭНКдЪмЬхБэДяЩЯЕї[68] ,ЖјIVаЭНКджазюжївЊЕФзїгУЮЛЕуМДЮЊRGDЖЬыФађСаЁЃвђДЫ,РћгУRGDзїЮЊАаЯђЛљЭХЕФдиЬхМДГЩЮЊИЮжзСівЉЮядиЬхбаОПЕФвЛИіаТЗНЯђЁЃ
BeljaarsЕШ[68]ЪзДЮНЋКЌRGDЦЌЖЯЕФЛЗыФ(C* GRGDSPC* ),СНЖЫАыызАБЫсвдЖўСђМќЯрСЌ)аоЪЮЕНШЫ бЊЧхАзЕААзБэУц(pCVI-HSA),ВЂПМВьСЫЦфдкИЮЯЫЮЌЛЏЖЏЮяФЃаЭжаЕФгІгУЁЃНсЙћЯдЪОга73%ЕФpCVI- HSAИЛМЏгкЯЫЮЌЛЏИЮдр;ЬхЭтЪЕбщвВБэУїpCVI-HSAЕФЧїИЮадРДдДгкЦфгыHSCsЯИАћЕФЬивьадНсКЯЁЃДЫКѓ,ЭѕМЊвЋПЮЬтзщгжЖдДЫЬхЯЕНјааСЫИФНј, ЫћУЧНЋЛЗыФжаЕФвЛИіАыызАБЫсИќЛЛЮЊРЕАБЫс (C* GRGDSPK* ),ДгЖјгУѕЃАЗМќДњЬцЫЋСђМќжЦБИЛЗRGDЖрыФ,ДѓДѓЬсИпСЫЛЗыФЕФЬхФкЮШЖЈад[69,70] ЁЃ
2.2.4 ЭИУїжЪЫсЪмЬхНщЕМЕФАаЯђИјвЉЯЕЭГ ЭИУїжЪЫс(HA)ЪЧвЛжжДѓЗжзгЬЧАЗОлЬЧ,ЪЧЙЙГЩЯИАћЭтЛљжЪКЭЯИАћМфжЪЕФжївЊГЩЗжЁЃЯИАћФЄБэУцгаЖржж HAЪмЬх,АќРЈCDЗжЛЏШК(Cluster determinant44, CD44)ЁЂИЮФкЦЄЯИАћЪмЬх(receptor-mediated endocytosis to liver endothelial cells, LEC)ЁЂЯИАћФкЭЬHAЪмЬх(HA receptor for endocytosis, HARE)ЁЂHAНщЕМЕФгЮЖЏадЪмЬх(receptor for hyaluronate-mediated motility, RHAMM)КЭСмАЭЙмФкЦЄЬивьадHAЪмЬх(lymphatic vessel endothelialhyaluronan receptor-1, (LYVE-1)ЕШ[71] ЁЃЦфжа,CD44дкжзСіЯИАћБэУцИпБэДя,LECжївЊДцдкгкИЮФкЦЄЯИАћ,HAREжївЊДцдкгкИЮаЧаЮЯИАћФк,вђДЫ,HAдкИЮАаЯђИјвЉЬхЯЕжаОпгаживЊЕФЮЛжУЁЃ
OhyaЕШ[72] баОПЗЂЯж,HAАќИВЕФОлРЕАБЫс-ОлШщЫсНКЪјЖдШЫИЮёМФкЦЄЯИАћОпгаУїЯдбЁдёад;ToriyaBeЕШ[73]вВЗЂЯжHAаоЪЮЕФжЌжЪЬхПЩБЛИЮёМФкЦЄЯИАћЭЬЪЩВЂдкИЮдрЯджјОлМЏ,ДЫИЮОлМЏЯжЯѓПЩБЛгЮРыHAвжжЦ,ЧвЦфОлМЏГЬЖШЫцжЌжЪЬхБэУцHAКЌСПдіМгЖјдіМгЁЃ
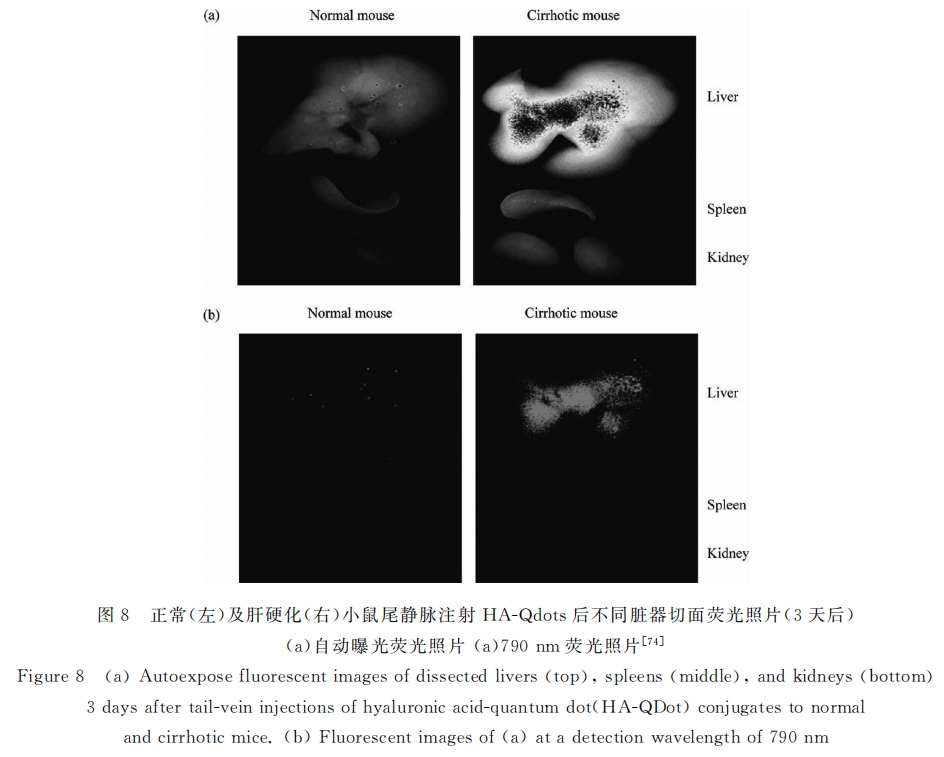
HahnПЮЬтзщРћгУHAЕФИЮАаЯђЙІФмПЊЗЂСЫвЛЯЕСаЯдгАМСЁЃШчKimЕШ[74] НЋHAСЌНгЕНСПзгЕуЩЯ(HA-Qdots)ВЂгІгУгкИЮгВЛЏМьВтЁЃНсЙћЯдЪОHAаоЪЮКѓ,ИЮЯИАћЖдСПзгЕуЕФАќЭЬФмСІДѓДѓЬсИп;ЖјЬхФкЪЕбщвВБэУїHA-QdotsПЩбИЫйИЛМЏгкЖЏЮяИЮдр(30min)ЁЃЬиБ№жЕЕУЙизЂЕФЪЧ:зЂЩф3ЬьКѓHA-QdotsЛљБОБЛе§ГЃаЁЪѓХХГіЬхЭт,ЖјдкИЮгВЛЏаЁЪѓИЮВПЛЙгаЯрЖдГЬЖШЕФДцСє(дМЮЊЗхжЕЕФ80%),МДЪЙЕНСЫ8ЬьКѓШдгадМ30%ЕФHA-QdotsзЄСєдкИЮдрВПЮЛ(ЭМ8)ЁЃЮЊСЫНЕЕЭСПзгЕуЕФЖОад,GohЕШ[75] жЦБИСЫHAаоЪЮФЩУзЬМСПзгЕу(HA-Cdot),ЭЈЙ§дкСПзгЕуБэУцАќИВФЩУзЬМЬсИпЦфЩњЮяЯрШнад,ЪЕбщНсЙћЯдЪОИУЬхЯЕШдОпгагХвьЕФИЮАаЯђФмСІЁЃ
2.2.5 бЊаЁАхЩњГЄвђзгЪмЬх-ІТНщЕМИЮАаЯђИјвЉЯЕЭГ бЊаЁАхЩњГЄвђзгЪмЬх(platete derived growth fact receptor,PDGFR)ЪЧвЛжжПчФЄЬЧЕААз,ОпгаРвАБЫсЕААзМЄУИЛюадЁЃPDGFRгЩСНжжбЧЕЅЮЛІСМАІТЙЙГЩ,ЦфЗжзгСПЮЊ170~180KDЁЃЖўепгыбЊаЁАхЩњГЄвђзг(PDGF)НсКЯСІЯрВюКмДѓ,ІСбЧЕЅЮЛгыPDGF-AСДМАPDGF-BСДгаНЯИпЕФЧзКЭСІ,ЖјІТбЧЕЅЮЛНігыPDGF-BСДгаИпЧзКЭСІЁЃОВжЙЕФHSCsБэУцжЛгаІСбЧЕЅЮЛ,HSCЛюЛЏКѓВХБэДяІТбЧЕЅЮЛ,ЖјЧввдІТбЧЕЅЮЛЮЊжїЁЃвђДЫ,дкЖЏЮяКЭШЫИЮЯЫЮЌЛЏЪБ,ИЮзщжЏжаPDGFR-ІТБэДяУїЯддіМг,ВЂгыHSCЪ§СПКЭИЮЯЫЮЌЛЏГЬЖШЯджјЯрЙи[76] ЁЃРћгУМЄЛюHSCsБэУцPDGFR-ІТИпБэДяЕФЬиад,баОПепПЊЗЂСЫвЛЯЕСаИЮАаЯђИјвЉЯЕЭГжЮСЦИЮЯЫЮЌЛЏМВВЁЁЃ
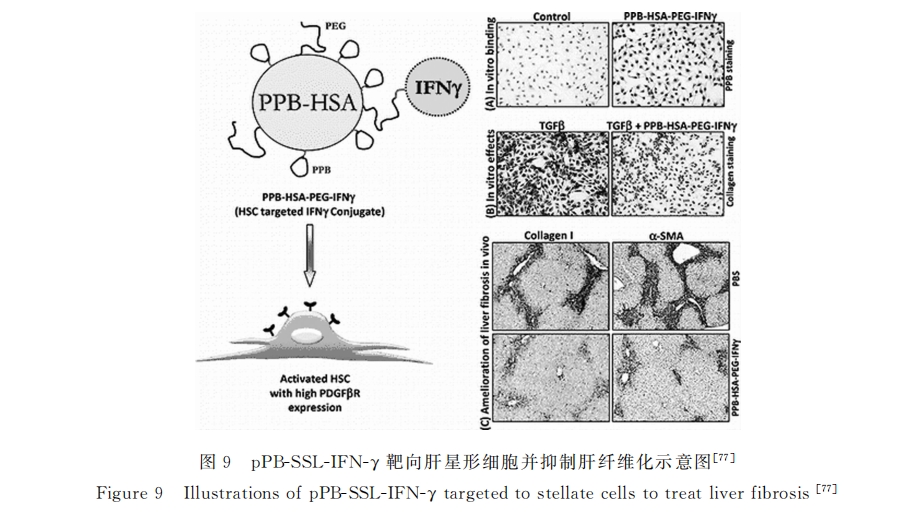
баОПБэУї:ЛЗыФC* SRNLIDC* (pPB)ЪЧPDGF-BBжаЕФжївЊЛюадЮЛЕу,ПЩгыPDGFR-ІТЗЂЩњЬивьадЕФНсКЯЁЃBansalЕШ[77,78]НЋPPBвдМАПЙЯЫЮЌЛЏвЉЮяИЩШХЫи)(IFN))ЭЌЪБСДНгЕНШЫбЊЧхАзЕААзБэУц(PPB-HAS-PEG-IFN))гУгкИЮЯЫЮЌЛЏЕФжЮСЦЁЃCCl4в§Ц№ЕФИЮЯЫЮЌЛЏЖЏЮяЬхФкЪЕбщЯдЪО:PPB-HAS-PEG-IFN)ПЩУїЯдИЛМЏгкЛюЛЏЕФHSCsжа;ЖјЧвЯрБШгЮРыЕФIFN),PPB-HAS-PEG-IFN)ПЩУїЯдМЄЛюВЂСзЫсЛЏаХКХзЊЕМгызЊТМЛюЛЏзг1(SATA1),ВЂвжжЦИЮЯЫЮЌЛЏЕФНјеЙ(МћЭМ9)ЁЃЭѕМЊвЋ[79]ПЮЬтзщРћгУPPBаоЪЮжЌжЪЬхАќдиIFN)(PPB-SSL-IFN-)),ЖЏЮяЬхФкЪЕбщЯдЪО:PPB-SSL-IFN-)жївЊИЛМЏгкИЮдр,ЖјЦфжа84.35%ЕФжЌжЪЬхМЏжагкМЄЛюЕФHSCsИННќЁЃ
Бэ2жаЖдВЛЭЌХфЬхНщЕМЕФЗЧЪЕжЪЯИАћАаЯђИјвЉЯЕЭГНјааСЫзмНс,ДгБэжаПЩМћ:ИЪТЖЬЧЁЂM6PвдМААаЯђIVНКдЪмЬхЕФRGDЛЗыФЖдИЮдрЕФАаЯђФмСІНЯЧП(ИЮРлЛ§ТЪвЛАудк60%~70%),ЖјHAвдМААаЯђPDGFЪмЬхЕФPPBЛЗыФЕФИЮАаЯђФмСІЯрЖдНЯШѕ(50%вдЯТ)ЁЃетгыХфЬхЕФаоЪЮУмЖШЫфШЛвВгавЛЖЈЙиЯЕ,ЕЋзмЬхЩЯРДПДгАЯьВЂВЛУїЯдЁЃЖдгкM6PЁЂRGDЛЗыФвдМАPPBЛЗыФНщЕМИјвЉЯЕЭГРДЫЕ,ЦфЯргІЪмЬхОљЗжВМгкHSCs,Чвдке§ГЃЬѕМўЯТЕЭБэДяЖјдкИЮЫ№ЩЫЕФЬѕМўЯТИпБэДяЁЃвђДЫЫфШЛИЮдрЕФВЁБфЖдЦфдкИЮдрВПЮЛзмЬхЕФИЛМЏГЬЖШУЛгагАЯь,ЕЋетаЉЪмЬхНщЕМЕФИјвЉЯЕЭГЖдАаЯИАћЕФАаЯђФмСІШдЬхЯжГіСЫУїЯдЕФИЮЫ№ЩЫвРРЕад,МДжЛдкИЮЯЫЮЌЛЏЧщПіЯТВХЖдАаЯИАћВњЩњЯджјЕФАаЯђзїгУЁЃгЩгкДѓЖрЪ§ИЮВЁЛМепЬхФкЛЗОГЖМЛсГіЯжвЛЖЈГЬЖШЕФИФБф,вђДЫетжжИЮЫ№ЩЫвРРЕЕФАаЯђадЖдгкИЮдрМВВЁЕФжЮСЦОпгаЪЎЗжЛ§МЋЕФвтвхЁЃДЫЭт,етжжаджЪЖдгкИЮЯЫЮЌЛЏ/ИЮгВЛЏЕФеяЖЯЭЌбљОпгаЪЎЗжживЊЕФзїгУ,ЮвУЧПЩвдЭЈЙ§АаЯђдиЬхдкИЮдржаЕФЗжВМЧщПіРДХаЖЯЪЧЗёГіЯжВЁБфЁЃ
2.3 ИЮжзСіЯИАћАаЯђ
РэЯыЕФЛЏСЦвЉЮяЕнЫЭЯЕЭГПЩЪЙвЉЮяНіНізїгУгкАЉБфЯИАћ,ЖјЖдЦфЫќе§ГЃЯИАћУЛгаЩЫКІЁЃвђДЫ,баОПепРћгУжзСіЯИАћКЭе§ГЃЯИАћМфЪмЬхБэДяЕФВювь,ПЊЗЂГівЛЯЕСажзСіЯИАћАаЯђИјвЉЯЕЭГ,ЪЙвЉЮяИЛМЏгкжзСізщжЏЁЃвдЯТ,ЮвУЧНЋМђЕЅНщЩмМИжждкИЮАЉжЮСЦжаГЃМћЕФжзСіЯИАћАаЯђИјвЉЯЕЭГЁЃ
2.3.1 ПЙЬхНщЕМЕФИЮАаЯђИјвЉЯЕЭГ ЕЅПЫТЁПЙЬх(monoclonal antiBodY, mAB),МђГЦЕЅПЙ , ЪЧНігЩвЛжжРраЭЕФЯИАћжЦдьГіРДЕФПЙЬхЁЃЕЅПЫТЁПЙЬхвдЦфЬивьадЧПЁЂДПЖШИпЁЂОљвЛадКУЕШгХЕу,вбЙуЗКгІгУгкЩњЮявНгУСьгђЁЃЕЅПЙПЩгыЗХЩфадЭЌЮЛЫиЁЂЛЏСЦвЉЮяМАЖОЫиЕШ,НсКЯаЮГЩУтвпХМСЊЮя(immunoconjugate),ЪЧФПЧАИЮдрМВВЁЕФжЮСЦЭООЖжЎвЛЁЃГЃбЁгУЕФЗХЩфадКЫЫига131IЁЂ125IЁЂ188ReЁЂ99mTcЕШ,вд131IзюЮЊГЃгУЁЃ2006Фъ,гЩГЩЖМЛЊЩёМЏЭХгыЕкЫФОќвНДѓбЇЕФГТжОФЯПЮЬтзщЙВЭЌбажЦЕФ[131I]2УРЭзЯЇЕЅПЙзЂЩфвКЛёЕУЙњМвЪГЦЗвЉЦЗМрЖНЙмРэОжАфЗЂЕФЩњВњЮФКХ(ЩЬвЕУћ:РћПЈЭЁ)ЁЃжЦМСЫљбЁгУЕФИЮАЉЕЅПЙУРЭзЯЇгыИЮАЉЯИАћБэУцЕФHAB18G/CD147ПЙдОпгаНЯЧПЧзКЭСІ,Цфв§ЕМ131IЗЂЩфИпФмІТСЃзгВЂЩБЩЫИЮАЉЯИАћ[80,81] ЁЃетЪЧФПЧАШЋЧђЮЈвЛвЛИігУгкжЮСЦдЗЂадИЮАЉЕФАаЯђвЉЮя,вВЪЧЮвЙњОпгазджїжЊЪЖВњШЈЕФПЙЬхРрвЉЮяЁЃ
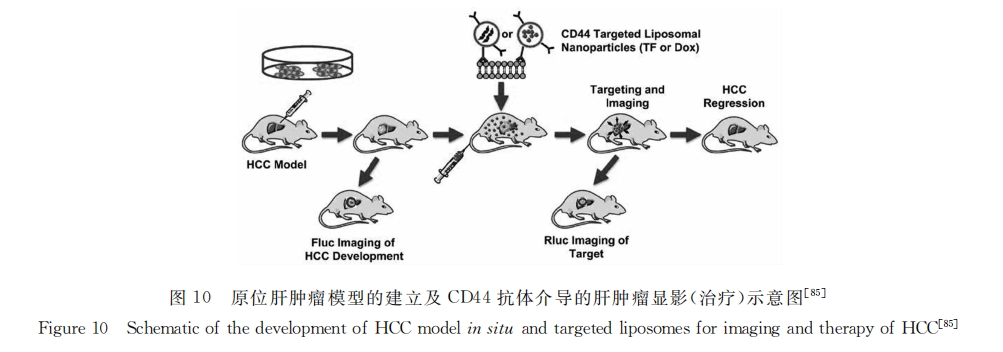
ДЫЭт,НЋЕЅПЙгыФЩУздиЬхВФСЯСЌНг,ПЩИГгшЮЂСЃЖдАаЯИАћЗжзгЫЎЦНЩЯЕФЪЖБ№ФмСІ,НЋЫљдивЉЮяДЋЫЭЕНжзСіЯИАћ,ДгЖјДѓДѓЬсИпвЉЮяСЦаЇ,МѕЧсЖОИБзїгУЁЃПЙЬхCD133ЁЂCD90ЁЂCD44ЁЂOV6ЁЂЩЯЦЄЯИАћ№ЄИНЗжзг(EPCAM)вдМАCD13ЕШ[82,83]гыЛЏСЦвЉЮяжЦГЩЕФжЌжЪЬхПЩЯджјЬсИпвЉЮяЕФАаЯђад,діЧПЛЏСЦаЇЙћЁЃШчЯђШйПЮЬтзщ[84]жЦБИСЫCD44ПЙЬхаоЪЮжЌжЪЬхФЩУзСЃзгВЂАќдиПЙАЉвЉЮяАЂУЙЫигУгкИЮАЉжЮСЦЁЃНсЙћЯдЪО,CD44ПЙЬхаоЪЮжЌжЪЬхПЩдкТуЪѓИЮдрВПЮЛОлМЏ,ЧвОпгаЯджјвжжЦжзСіЩњГЄЕФаЇЙћЁЃ
ОЁЙмЕЅПЫТЁПЙЬхжЦМСвбОШЁЕУРэЯыЕФГЩЙћ,ЖржжВњЦЗвбОЩЯЪа,ЕЋШдШЛгааэЖржЕЕУНтОіЕФЮЪЬтЁЃШчЪѓдДадПЙЬхгІгУгкШЫЬхЭљЭљЛсВњЩњШЫПЙЪѓПЙЬх(Human anti-mouse antiBodied, HAMA),ДгЖјв§Ц№Й§УєЗДгІ,ЯожЦЪѓдДадПЙЬхдкСйДВЩЯЕФгІгУ[85,86] ;ЖјФПЧАЩЯЪаЕФПЙЬхжЦМСжаЭъШЋШЫдДЛЏЕФЕЅПЙЯрЖдНЯЩй,ЖрДЮжиИДЪЙгУПЩФмв§Ц№УтвпдадКЭЦфЫќЮЃКІ;ПЙЬхЕФаЁаЭЛЏ,ПЙЬхМАЦфХМСЊЮяОљЮЊДѓЗжзгЮяжЪ,ХгДѓЕФЗжзгФбвдЭЈЙ§УЋЯИЙмФкЦЄВуКЭЯИАћЭтМфЯЖЕНДяЪЕЬхСіЩюВПЕФжзСіЯИАћ;ДЫЭт,ПЙЬхвЉЮяДѓЖрАКЙѓ,ШчРћПЈЭЁашвЊдМ2.88ЭђдЊ/жЇ,ЦеЭЈВЁШЫФбвджЇИЖЕШЕШЁЃ
2.3.2 вШЕКЫиЪмЬхНщЕМЕФАаЯђИјвЉЯЕЭГ вШЕКЫиЪмЬх(Insulin Receptor)НщЕМЕФИЮАаЯђИјвЉЯЕЭГбаОПЪМгкЩЯЪРМЭ70ФъДњ,ДѓСПбаОПБэУїИЮАЉЯИАћФЄЩЯвШЕКЫиЪмЬхЕФУмЖШКЭЧзКЭСІОљНЯе§ГЃИЮЯИАћгаЫљдіМг[87,88] ЁЃШчkurtaranЕШ[88] ЕФбаОПНсЙћЯдЪОШЫИЮАЉЯИАћЕФвШЕКЫиЪмЬхУмЖШЮЊе§ГЃЯИАћЕФ1000БЖЁЃХЗЯўКьЕШ[89]вВЗЂЯжH22ИЮАЉЯИАћНЯе§ГЃаЁЪѓИЮЯИАћГЌСПБэДявШЕКЫиЪмЬхЁЃДЫЭт,гыЦфЫќаЁЗжзгХфЛљЯрБШ,вШЕКЫигаНЯЖрЛюадЮЛЕу,взгкгыаЁЗжзгвЉЮяЁЂЕААзЁЂПЙЬхЛђЗХЩфадКЫЫиЕШЗЂЩњХМСЊЁЃЛљгкЩЯЪібаОП,ПЩНЋвШЕКЫизїЮЊИЮАЉжїЖЏАаЯђИјвЉЯЕЭГЕФЁАЕЏЭЗЁБ,ЭЈЙ§ЪмЬхНщЕМЗНЪННЋвЉЮяЖЈЯђзЊдЫВЂЕМШыИЮАЉЯИАћ,ДгЖјЬсИпвЉЮяЩњЮяРћгУЖШЁЂМѕЩйШЋЩэЖОИБзїгУЁЃ
LiuЕШ[90] вдвШЕКЫиЮЊЕМЯђХфЛљ,УзЭаньѕЋЮЊФЃаЭПЙжзСівЉЮя,жЦБИСЫУзЭаньѕЋ-вШЕКЫиХМСЊЮя,ВЩгУКЩСіИЮАЉаЁЪѓЮЊжзСіФЃаЭ,ЯЕЭГЕибаОПСЫвШЕКЫиЪмЬхНщЕМЕФИЮАЉЯИАћжїЖЏАаЯђИјвЉЯЕЭГЕФаджЪЁЃвЉЮяЖЏСІбЇбаОПНсЙћЯдЪО,ХМСЊЮяОпгаНЯИпЕФжзСіАаЯђадКЭНЯГЄЕФбЊвКбЛЗЪБМфЁЃЬхЭтвЉаЇбЇЦРМлНсЙћБэУї,гыдСЯвЉЯрБШХМСЊЮяФмИќбИЫйЕиНјШыжзСіЯИАћВЂЪЭЗХвЉЮя,ВЂЧвЪЭЗХГіЕФвЉЮяШдБЃГжвжСіЛюад,ДгЖјГфЗжБЃжЄХМСЊЮяЕФИЮАаЯђвжСіаЇЙћ;ЭЌЪБХМСЊЮяЖде§ГЃИЮЯИАћЕФЖОадвЊУїЯдЕЭгкдвЉ,ЫЕУїЦфФмбЁдёадЕиЩБЩЫИЮАЉЯИАћ,НЕЕЭвЉЮяЖде§ГЃИЮЯИАћЕФЫ№ЩЫЁЃХЗЯўКьЕШ[91]жБНгНЋЕтЭббѕФђмеаоЪЮЕНвШЕКЫи(insulin-IUdR)ЩЯЖдИЮАЉНјааАаЯђжЮСЦ,ЪЕбщНсЙћБэУїinsulin-IUdRгыШЫИЮАЉЯИАћЭЌбљОпгаНЯИпЕФЧзКЭФмСІ,ЪЧгыСкНќе§ГЃИЮЯИАћЧзКЭФмСІЕФ1.45БЖЁЃЛЦОъ[92] вдвШЕКЫиЮЊдиЬх,Аќди5-ЗњФђрзрЄ,жЦБИСЫвШЕКЫи-5-ЗњФђрзрЄХМСЊЮя,НЈСЂСЫаЁЪѓH22ИЮАЉЪЕЬхСіФЃаЭ,ЖдХМСЊЮядкКЩСіаЁЪѓЬхФкЗжВМЁЂвЉДњЖЏСІбЇКЭХМСЊЮяАаЯђадНјааСЫбаОП,ВЂгыгЮРы5-ЗњФђрзрЄНјааСЫЖдБШЗжЮі,НсЙћЯдЪО,ХМСЊЮяЖдH22ИЮАЉЪЕЬхСігаУїЯдЕФАаЯђад,ХМСЊЮяНЯдвЉЕФвжСізїгУИќПь,ФмЙЛЗЂЛг5-FuЕФПЙАЉЛюадзїгУ,ФмДяЕНИЮАаЯђПЙАЉвЉЮяЕФвЊЧѓЁЃ
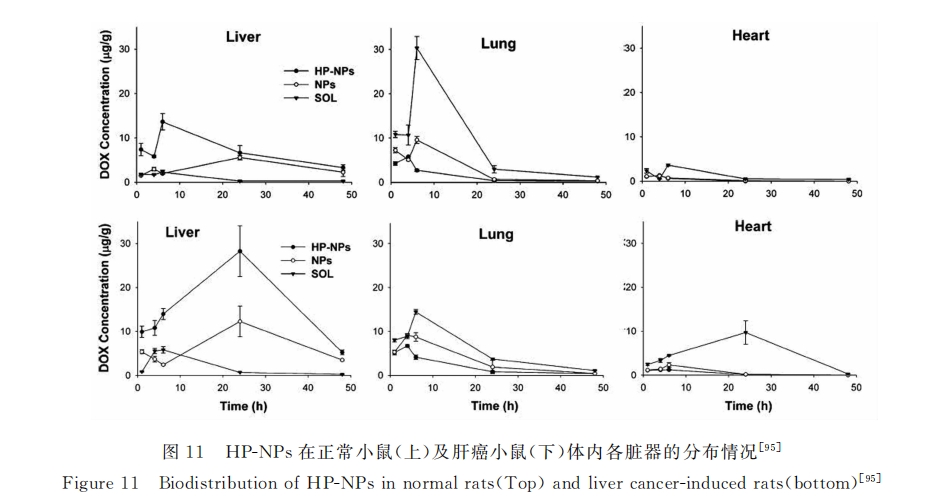
2.3.3 ЦфЫќАаЯђИјвЉЬхЯЕ ЕЭУмЖШжЌЕААзЪмЬхНщЕМ габаОПЯдЪО:ЕЭУмЖШжЌЕААзЪмЬхдкИЮжзСіЯИАћжагаНЯИпБэДя[93] ЁЃChang ЕШ[94] РћгУбЊпВпј(Hematoporphyrin,HP)гыLDLЪмЬхМфЬивьЯрЛЅзїгУ,жЦБИСЫHPаоЪЮАќдиDOXЕФХЃбЊЧхАзЕААз(BSA)ФЩУзПХСЃ(HP-NPs)ЁЃНсЙћЯдЪО:HepG2ЯИАћЖдHP-NPs ЕФАќЭЬФмСІУїЯдЧПгкЮДаоЪЮФЩУзСЃзг(NPs);ЖјИќЮЊживЊЕФЪЧ,HP-NPsдкЛМИЮАЉаЁЪѓИЮФкРлЛ§ГЬЖШЯджјИпгке§ГЃаЁЪѓЕФИЮФкРлЛ§ГЬЖШ(AUCжТАЉаЁЪѓИЮ :AUCе§ГЃаЁЪѓИЮ =2.45),ЯдЪОГіЦфдкИЮжзСіжЮСЦжаЕФЖРЬигХЪЦ(ЭМ11)ЁЃ
3-аЭСзжЌѕЃМЁДМЕААзОлЬЧНщЕМ
3-аЭСзжЌѕЃМЁДМЕААзОлЬЧ(GPC-3)ЪЧвЛжжЯИАћФЄЕААз,ЦфдкИЮАЉЯИАћБэУцИпБэДя;ЖјЧвGPC-3дкбЊ вКжаВЂВЛДцдк,жЛдкЯИАћФЄБэУцБэДя,вђДЫЗЧГЃЪЪКЯгУгкжзСіАаЯђжЮСЦЁЃLeeЕШ[95]ЭЈЙ§гЋЙтУтвпЕФЗНЗЈ дкЬхЭтЩИбЁСЫGPC-3ХфЬхЖрыФ,НсЙћЯдЪООпгаTyr-Phe-Leu-Thr-Thr-Arg-GlnађСаЕФЖрыФПЩгыGPC-3ИпБэДяЕФHepG2ЯИАћгаНЯЧПЕФНсКЯ,гаЭћГЩЮЊаТаЭИЮАаЯђХфЬхЁЃ
ЖрыФЪмЬхНщЕМ
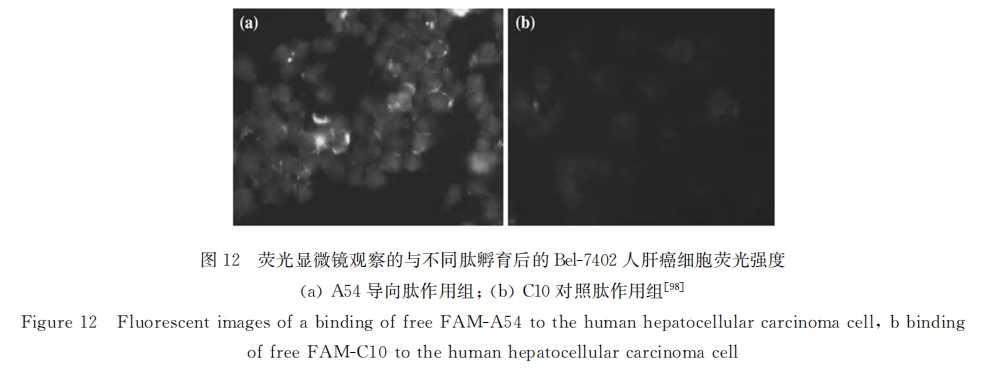
ЖјЧЎFПЮЬтзщвВРћгУЪЩОњЬхыФПтММЪѕзджїЩИбЁГіОпгаЬивьАаЯђBel-7402ИЮАЉЯИАћЕФЕМЯђыФ AGKGTPSLETTP(A54),ЭЈЙ§ЙВОлНЙГЩЯёЯдЮЂОЕЙлВьСНепМфЧзКЭСІ,ЗЂЯжОA54ЕМЯђыФЗѕг§КѓЕФ Bel-7402ЯИАћГЪЯжУїЯдгЋЙт(МћЭМ12),ЖјгыЖдееыФC10Зѕг§ЕФBel-7402гЋЙтМЋШѕ(МћЭМ1.8B)[96,97] ЁЃНјвЛВНЕФбаОПБэУї,A54гыбѕЛЏЬњДХадФЩУзСЃ(SIONs)ХМСЊКѓПЩвдЕУЕНОпгаМцОпИЮАаЯђадКЭДХЫЋжиЯь гІадЕФФЩУзСЃ,ФЩУзСЃдкЭтМгДХГЁзїгУЯТдкИЮдржзСіВПЮЛИпЖШИЛМЏ[98,99] ,гаЭћзїЮЊвЛРраТаЭЕФИЮАаЯђВФСЯЁЃ
ДЫЭт,ГЃМћЕФжзСіЯИАћАаЯђХфЬхОљПЩгУгкИЮжзСіЯИАћАаЯђ,ШчвЖЫс[100]ЁЂзЊЬњЕААз[101,102] вдМАећКЯЫиХфЬх(RGDЖрыФ)[103,104]ЕШНщЕМЕФОљдкИЮАЉМьВтМАжЮСЦСьгђгазХЙуЗКгІгУ,етаЉФкШндкKolhatkarЕШ[105] ЕФзлЪіжаОљгаЯъЯИНщЩм,дкДЫОЭВЛЯъЯИзИЪіСЫЁЃЕЋЮвУЧгІИУзЂвтЕНвЛИіЮЪЬт,дкжзСіЯИАћБэУцИпБэДяЕФЪмЬхЭљЭљЭЌЪБДцдкгкЦфЫќзщжЏжа;ЖјЯрЖде§ГЃзщжЏРДЫЕ,жзСізщжЏЯИАћЪ§СПгжЯрЖдНЯЩй,вђДЫЦфЖдвЉЮяЕФИЛМЏзїгУгаЯо,ДѓВПЗжвЉЮяЛЙЪЧдкЬхФкЦфЫќЦїЙйЗжВМ,вЉЮяРћгУТЪЬсЩ§гаЯоЁЃШчЛЦОъЕШбаОПЯдЪО:ЯрЖдЦеЭЈ5-ЗњФђрзрЄ(5-Fu)вШЕКЫиаоЪЮЕФ5-Fu(Ins-5-Fu)дкжзСіВПЮЛЕФИЛМЏГЬЖШгаКмДѓЬсЩ§(дМЬсИп54.7%),ЕЋЦфдкжзСіВПЮЛЕФСПШдНіеМзЂЩфзмСПЕФ1%зѓгвЁЃвђДЫзїепШЯЮЊ:ЮЊСЫЬсИпвЉЮяРћгУТЪ,ЪзЯШгІИГгшвЉЮядиЬхвЛЖЈЦїЙйАаЯђФмСІ(МДвЉЮяЪзЯШИЛМЏгкИЮдр),дкДЫЛљДЁЩЯдйРћгУдиЬхЕФВЁБфЯИАћбЁдёад,НЋвЉЮяНјвЛВНИЛМЏгкВЁБфзщжЏ,етбљВХФмДяЕНИќКУЕФжЮСЦаЇЙћЁЃ
3 еЙЭћ
НќФъРД,ЫцзХжзСібЇЁЂВЁРэбЇЁЂЗжзгЩњЮябЇвдМАВФСЯбЇЕФЗЂеЙ,ШЫУЧЖдИЮдрМВВЁЕФЗЂВЁЛњРэ,ИЮЙІФмЕФЗжзгЛњжЦЕФШЯЪЖгаСЫжЪЕФЬсЩ§,ЖјЛљгкФЩУзВФСЯЕФИЮАаЯђИјвЉЯЕЭГЕФЗЂеЙвВгаСЫГЄзуЕФНјВНЁЃБОЮФЖдгкНќФъРДеыЖдИїРрИЮдрМВВЁжЮСЦЫљПЊЗЂЕФжї/БЛЖЏдивЉ(Лљвђ)ЬхЯЕНјааСЫзлЪіЁЃДгБОЮФзмНсжаПЩМћ:ЛљгкВЛЭЌИЮдрМВВЁЕФЗЂВЁЛњРэ,баОПепУЧеыЖдИЮЪЕжЪЯИАћМАЗЧЪЕжЪЯИАћБэУцВЛЭЌЪмЬхЩшМЦВЂжЦБИСЫЖржжИЮАаЯђИјвЉ/диЛљвђЬхЯЕЁЃетаЉЬхЯЕдкЯИАћЫЎЦНМАЖЏЮяЫЎЦНОљБэЯжГіСЫгХвьЕФИЮзщжЏ/ИЮЯИАћАаЯђФмСІ,дкжЮСЦжавВДѓДѓЕФЬсИпСЫвЉЮя/ЛљвђЕФжЮСЦЛђзЊШОаЇЙћЁЂНЕЕЭСЫЯИАћЖОадМАИБзїгУ,ЩѕжСЖдгкЖрвЉФЭвЉадвВгавЛЖЈЕФвжжЦаЇЙћ,ЯдЪОГіСЫНЯКУЕФгІгУЧАОАЁЃдкетРягІИУЧПЕїЕФвЛЕуЪЧ:ИЮдрМВВЁЕФЗЂВЁЩцМАЖрЗНУцвђЫи,ЛњРэвВЪЎЗжИДдг,гааЉМВВЁЫфШЛЗЂВЁдкЪЕжЪЯИАћЩЯ,ЕЋвВКЭЦфЫќИЮВПЯИАћгазХМЋЦфживЊЕФСЊЯЕЁЃШчдЗЂадИЮАЉвЛАуЮЛгкИЮЪЕжЪЯИАћ,ЕЋЦфЭљЭљгЩВЁЖОадИЮбзЁЂИЮЯЫЮЌЛЏЁЂИЮгВЛЏЗЂеЙЖјРД,ЖјетаЉМВВЁЭљЭљгыKCsЯИАћЁЂHSCsЯИАћвдМАSECsЯИАћЕШЗЧЪЕжЪЯИАћЙиЯЕУмЧаЁЃвђДЫ,ЖдгкИЮдрМВВЁЕФжЮСЦ,вЊзлКЯПМТЧИЮЪЕжЪЯИАћКЭЗЧЪЕжЪЯИАћЕФАаЯђзїгУЁЃ
СэЭтЮвУЧвВгІИУПДЕН,Г§жЌжЪЬхжЎЭтФПЧАЩйгаФЩУзИјвЉЬхЯЕзпШыСйДВЪЙгУ;ЖјЕНФПЧАЮЊжЙШдУЛгаШЮКЮвЛжжФЩУзАаЯђИјвЉЯЕЭГНјШыСйДВЪдбщЁЃетЪЧЖрЗНУцдвђдьГЩЕФ,БШШч:(1)ФПЧАбаОПЕФЖрЪ§АаЯђжЦМСОљЮЊОВТізЂЩфИјвЉ,ЖјДЫжжИјвЉЗНЪНЖдгкаТВФСЯЕФбЊвКЯрШнадМАЩњЮяЯрШнадЕФвЊЧѓНЯИп,вђДЫКмФбЛёЕУЩѓХњ;(2)АаЯђВФСЯ,ЭљЭљашвЊНЋАаЯђХфЬхЭЈЙ§ЛЏбЇМќаоЪЮЕНгаЛњ/ИпЗжзгВФСЯЩЯ,ДгЖјдіМгСЫВФСЯЕФжЦБИГЩБОвдМАВњЦЗжЪСПЕФПижЦФбЖШ;(3)КмЖрИЮАаЯђИјвЉЬхЯЕЕФАаЯђЛњРэМАДњаЛЭООЖШдВЛЧхГў,ШчИЪВнЫс/ИЪВнДЮЫсЪмЬхЕФжжРржСНёШдЮДШЗШЯЁЃетаЉЮЪЬташвЊВФСЯбЇМвЁЂЗжзгЩњЮябЇМввдМАСйДВвНЩњКЯзїНтОіЁЃ
Г§ДЫжЎЭт,ЮвУЧЛЙгІзЂвтЕН:РэЯыЕФИЮАаЯђИјвЉЯЕЭГВЛНігІдкНЋвЉЮяИЛМЏВЂзЄСєдкИЮдрВПЮЛ,ЖјЧвгІОЁПЩФмЬсИпЦфдкВЁБфЯИАћЕБжа(ШчИЮАЉЯИАћ)ЕФХЈЖШ,ДгЖјМѕЩйЖде§ГЃЯИАћЕФЩЫКІЁЃетВЛНівЊЧѓАаЯђИјвЉЯЕЭГОпгаИЮАаЯђФмСІ,ЛЙгІОпБИВЁБфЯИАћЪЖБ№ЙІФмЁЃБэ3жаСаОйСЫвЛаЉОпгаВЁБфЯИАћЪЖБ№ФмСІЕФХфЬх,етаЉХфЬхНщЕМЕФИЮАаЯђИјвЉЯЕЭГгаЭћдкЮДРДЕФИЮдрМВВЁжЮСЦжаЗЂЛгИќДѓЕФзїгУЁЃСэЭт,дкИЮАаЯђвЉЮяЕнЫЭЕФЛљДЁЩЯИГгшдиЬхвЛЖЈЛЗОГУєИаЙІФмгаПЩФмГЩЮЊНтОіетвЛЮЪЬтЕФгааЇЭООЖЁЃШчБОзщНќРДбаЗЂСЫМцОпИЮАаЯђКЭЫсУєИаЙІФмЕФаТаЭИЮАаЯђИјвЉ/ЪЭвЉЬхЯЕ,ЫќВЛНіПЩвдНЋПЙАЉвЉЮяАЂУЙЫиИЛМЏгкИЮдр,ЖјЧвПЩвдбЁдёадЕФдкжзСіВПЮЛ(PH6.5зѓгв)НЋвЉЮяЪЭЗХГіРД,ДгЖјМѕЩйЖде§ГЃИЮзщжЏ(PH7.4зѓгв)ЕФЩЫКІЁЂЬсИпвЉЮяАВШЋадМАЩњЮяРћгУТЪЁЃ
СэЭт,ЮвУЧЭЈЙ§ЖдБШЗЂЯжСЫвЛИіЯжЯѓ:АаЯђИјвЉЯЕЭГЫфШЛПЩвдНЋвЉЮяУїЯдИЛМЏгкИЮдр(вЛАуПЩЮЊЖдеезщЕФ1.5БЖзѓгв),ШЛЖјвЉЮяЕФСЦаЇВЂЮДЬхЯжГіЯргІЕФЯджјБфЛЏ(IC50жЕДѓЖрБфЛЏВЛДѓ)ЁЃЮвУЧШЯЮЊетжївЊгавдЯТМИЗНУцдвђ:ЪзЯШЪЧвЉЮяЕФЪЭЗХЫйЖШЁЃЯрЖдДЋЭГЕФвЉЮяШмвК(freedrug)ФЩУздиЬхжаЕФвЉЮяЭљЭљашвЊвЛЖЈЕФЪБМфВХФмЭъШЋЪЭЗХГіРД,етОЭЪЙЕУФЩУзИјвЉЬхЯЕдкжЮСЦЕФЦ№ЪМНзЖЮгааЇвЉЮяХЈЖШУїЯдЕЭгквЉЮяШмвК,ДгЖјЪЙЕУЦфБэЙлЯИАћЖОадЯТНЕЁЃСэЭт,ФЩУздивЉЬхЯЕНјШыЯИАћКѓЪзЯШЛсНјШыФкКЬх(PH5~6),вЉЮядкДЫЪЭЗХКѓЭљЭљЛсБЛФкКЬхжаДцдкЕФИїжжИпХЈЖШСбНтУИЗжНтДгЖјдкдкБэЙлЩЯЯдЪОЮЊвЉаЇЯТНЕЁЃвђДЫ,ЮЊСЫБЃГжвЉаЇБиаыЪЙФЩУзЬхЯЕОЁПьРыПЊФкКЬх,МДФкКЬхЬгвнЁЃДЫЭт,Г§СЫПМТЧВФСЯгыШЫЬхЛЗОГЕФгАЯьжЎЭт,ЮвУЧдкЩшМЦАаЯђИјвЉЯЕЭГЪБЛЙгІзЂвтвЉРэбЇЗНУцЕФвђЫиЁЃШчзЯЩМДМПЩгеЕМКЭДйНјЮЂЙмЕААзОлКЯ,ДгЖјвжжЦСЫЯИАћЗжСбКЭдіжГ,ЗЂЛгПЙжзСізїгУ,вђДЫБиаыНЋзЯЩМДМЪЭЗХЕНЯИАћжЪжаВХФмЦ№ЕНСЦаЇ;ЖјАЂУЙЫижївЊЭЈЙ§ЧЖШыDNAЖјвжжЦКЫЫсЕФКЯГЩДгЖјЦ№ЕНжЮАЉЙІФм,вђДЫаыНЋЦфЪфЫЭжСЯИАћКЫВХФме§ГЃЦ№аЇ,етаЉЖМЩцМАЕНСЫИќЩюВуДЮЕФАаЯђФмСІЁЊЁЊЁЊбЧЯИАћАаЯђЛђЯИАћЦїАаЯђЁЃетаЉгаД§гкЗжзгЩњЮябЇвдМАВФСЯбЇЕФНјвЛВНЗЂеЙВХФмгааЇНтОіЁЃ
Утд№ЩљУїЃКБОЮФЮЊаавЕНЛСїбЇЯАЃЌАцШЈЙщдзїепМАддгжОЫљгаЃЌШчгаЧжШЈЃЌПЩСЊЯЕЩОГ§ЁЃЮФеТБъзЂгазїепМАЮФеТГіДІЃЌШчашдФЖСдЮФМАВЮПМЮФЯзЃЌПЩдФЖСддгжОЁЃ
