
’Σ“ΣΘΚΙ«ΦήΜΖκΡ «œΏ–‘κΡΒΡCΕΥΚΆNΕΥΆ®ΙΐθΘΑΖΦϋΫχ–– ΉΈ≤ΜΖΚœΕχ–Έ≥…ΒΡΜΖΉ¥Ζ÷Ή”. ―–ΨΩ’Ώ¥”œΗΨζΓΔ’φΨζΓΔ÷≤ΈοΚΆΕ·Έο÷–ΖΔœ÷ΝΥ¥σΝΩΒΡΙ«ΦήΜΖκΡ. ’β÷÷ ΉΈ≤ΜΖΚœΒΡΫαΙΙΘ§ ΙΒΟΙ«ΦήΜΖκΡΨΏ”–ΚήΚΟΒΡΟΗΈ»Ε®–‘ΓΔ»»Έ»Ε®–‘ΚΆΜ·―ßΈ»Ε®–‘Θ§≤ΩΖ÷Ι«ΦήΜΖκΡΨΏ”–œΗΑϊΡΛΆ®ΆΗ–‘. Ι«ΦήΜΖκΡΖ÷Ή”“λ≥ΘΒΡΈ»Ε®–‘ΚΆΗΏ–ßΒΡ…ζΈοΜν–‘Θ§ ΙΒΟΤδ≥…ΈΣΡΩ«Α“©ΈοΝλ”ρΒΡ―–ΨΩ»»Βψ. ΈΣΝΥΗϋ…ν»κΒΊ―–ΨΩΥϋΟ«ΒΡΫαΙΙΚΆΙΠΡήΘ§Ι«ΦήΜΖκΡΒΡ÷Τ±Η≥…ΈΣ“ΜΗω÷Ί“ΣΈ Χβ. Η≈ ωΝΥΜ·―ßΚœ≥…Ι«ΦήΜΖκΡΒΡ“Μ–©ΖΫΖ®Θ§Αϋά®ΘΚΘ®1Θ©ΙΧœύΜΖΚœ≤Ώ¬‘ΘΜΘ®2Θ©“ΚœύΜΖΚœ≤Ώ¬‘ΘΜΘ®3Θ©Ζ÷Ή”ΡΎΉ‘»ΜΜ·―ßΝ§Ϋ”≤Ώ¬‘Θ§≤ΔΕ‘’β–©ΖΫΖ®ΒΡΧΊΒψΚΆ–߬ Ϋχ––ΝΥΧ÷¬έ±»Ϋœ.
Ι«ΦήΜΖκΡ(Backbone cyclized peptides) «œΏ–‘ΕύκΡΒΡΆΖΈ≤Ά®ΙΐθΘΑΖΦϋœύΝ§Εχ–Έ≥…ΒΡΜΖΉ¥Ζ÷Ή”. ”κœΏ–‘ΕύκΡœύ±», Ι«ΦήΜΖκΡΨΏ”–ΫœΈΣΗ’–‘ΒΡΜΖΙ«ΦήΫαΙΙΧΊ’ς, “ρΕχΕ‘ΒΑΑΉΥ°ΫβΟΗΈ»Ε®–‘œ‘÷χΧαΗΏ, …θ÷Ν≤ΩΖ÷Ι«ΦήΜΖκΡΨΏ”–ΕάΧΊΒΡœΗΑϊΡΛΆ®ΆΗ–‘. ’ΐ «’β“Μ‘≠“ρ, Ι«ΦήΜΖκΡ≥…ΈΣΒ±ΫώΙζΦ “©ΈοΝλ”ρΒΡ÷Ί“Σ―–ΨΩΕ‘œσ[1ΓΪ7]. ‘γ‘ΎΒΎΕΰ¥Έ άΫγ¥σ’ΫΤΎΦδ, Υ’ΝΣΩΤ―ßΦ“ΗΏΥΙΨΆΖΔœ÷”… 10 ΗωΑ±ΜυΥαΉι≥…ΆΖΈ≤ΜΖΚœΕχ≥…ΒΡΕΧΗΥΨζκΡΨΏ”–“÷÷ΤœΗΨζ…ζ≥ΛΒΡΙΠΡή[8]. Υϋ «Εΰ’ΫΤΎΦδ÷ΈΝΤ…Υ≤Γ»Υ…ΥΩΎΗ–»ΨΒΡ÷ς“Σ“©Έο, ΆλΨ»ΝΥΈό ΐ»ΥΒΡ…ζΟϋ. ΕΧΗΥΨζκΡΒΡΖΔœ÷≥…ΈΣΜΖκΡ―–ΨΩΒΡ÷Ί“Σάζ–‘ ¬Φΰ. ΥφΚσ, ―–ΨΩ’Ώ‘ΎœΗΨζΓΔ’φΨζΓΔ÷≤ΈοΚΆΕ·Έο÷–ΨυΖΔœ÷ΜΖκΡ, Τδ÷––μΕύ≤ΜΖΠ“©ΈοΙΠΡή. άΐ»γ, ”… 11 ΗωΑ±ΜυΥαΉι≥…ΒΡΜΖφΏΥΊΩ…”Ο”ΎΤςΙΌ“Τ÷≤÷–Ζά÷ΙΟβ“ΏΖ¥”Π; Κ§”–ΕύΗωΜΖΉ¥ΫαΙΙΒΡ»ιΥαΝ¥«ρΨζκΡΩ…”Ο”ΎΖά÷Ι ≥ΤΖΗ·Αή; Εύ’≥ΥΊΩ…”Ο”Ύ÷ΈΝΤ≥ΠΈΗΦ≤≤ΓΒ»[9].
Ι«ΦήΜΖκΡΒΡΧλ»ΜΙΠΡή «Υό÷ςΖά”υΉς”Ο, Φ¥Ά®Ιΐ“÷÷ΤΆβά¥…ζΈοΒΡ…ζ≥Λά¥±ΘΜΛ±ΨΈο÷÷ΒΡ…ζ¥φΚΆ―”–χ. Ά®ΙΐΜν–‘ Β―ι, ―–ΨΩ’ΏΖΔœ÷Ι«ΦήΜΖκΡΜΙΨΏ”–“Μ–©ΕάΧΊ…ζΈοΙΠΡή,Ω…”Ο”Ύ÷ΈΝΤ»ΥάύΦ≤≤Γ. »γ¥φ‘Ύ”Ύ÷≤Έο÷–ΒΡΜΖΕύκΡ(Cyclotide)ΨΏ”–÷ζΉ”Ι§ ’ΥθΜν–‘ΓΔΩΙΨζ–‘ΓΔΩΙ HIV Μν–‘“‘ΦΑΩΙ÷ΉΝωΜν–‘[10]. »ΜΕχ, ¥”…ζΈο÷–ΒΟΒΫΒΡΙ«ΦήΜΖκΡΩ…ΡήΕ‘»ΥΧε≤ζ…ζ…ζΈοΕΨ–‘, “ρ¥ΥΕύ ΐ≤ΜΡή÷±Ϋ””Ο”ΎΝΌ¥≤ ‘―ι , Εχ–η“ΣΕ‘ΤδΫαΙΙΫχ––ΗΡ‘λ . άΐ »γ , ΈΣΝΥΫΒΒΆCyclotide ΒΡ…ζΈοΕΨ–‘, ―–ΨΩ’Ώ–η“ΣΕ‘ΤδΗω±πΈΜΒψΫχ––ΆΜ±δΜρ’ΏΆ®ΙΐΓΑΦόΫ”Γ±ΒΡΖΫ ΫΫΪΨΏ”–…ζΈοΜν–‘ΒΡΆβ‘¥ΙΠΡήκΡ»ΎΚœ”Ύ Cyclotide ÷–[11].
Ι«ΦήΜΖκΡΩ…“‘Ά®Ιΐ 3 ÷÷ΆΨΨΕΜώΒΟ: ¥”…ζΈοΧε÷–Ζ÷άκΓΔΜυ“ρ÷ΊΉιΦΦ θΚΆΜ·―ߥ”ΆΖΚœ≥…. ¥”…ζΈοΧεΡΎ¥ΩΜ·Ι«ΦήΜΖκΡΒΡΖΫΖ®¥φ‘ΎΖ÷άκΡ―Ε»¥σΓΔ–ß¬ ΒΆΚΆΙΛΉςΝΩ¥σΒΡ»±Βψ. ”…”ΎΙ«ΦήΜΖκΡ¥φ‘ΎΧΊ βΒΡΆΖΈ≤ΜΖΚœΒΡθΘΑΖΦϋΫαΙΙ, ¥ΪΆ≥Μυ“ρ÷ΊΉι±μ¥οΦΦ θ“≤≤ΜΡή÷±Ϋ””Ο”ΎΙ«ΦήΜΖκΡΒΡ÷Τ±Η, –η“ΣΕ‘Ψζ÷÷Ϋχ––‘ΌΗΡ‘λ, “ρΕχΗΟΖΫΖ®ΦΦ θΡ―Ε»œύΕ‘ΫœΗΏ. Ι«ΦήΜΖκΡΕ‘Ψζ÷÷ΒΡΕΨ–‘“≤Μα≥Θ≥ΘΒΦ÷¬Μυ“ρ÷ΊΉι±μ¥ο ßΑή. ¥ΥΆβ, …ζΈο±μ¥οΚ§”–Ζ«Χλ»Μ–ό ΈΒΡΙ«ΦήΜΖκΡΒΡΡήΝΠ °Ζ÷”–œό. œύ±»«ΑΝΫ÷÷ΖΫΖ®, Μ·―ߥ”ΆΖΚœ≥…Ι«ΦήΜΖκΡΨΏ”–”≈ Τ, »γΩ…“‘»Έ“βΗΡ±δΑ±ΜυΥαΒΡ–ρΝ–, Ω…“‘ Βœ÷ΈΜΒψΒΡ»Έ“βΆΜ±δ, “‘ΦΑΩ…“‘»Έ“βΖ«Χλ»ΜΑ±ΜυΥαΒΡ«Ε»κΒ».
¥”ΜΖΜ·Ζ¥”ΠΒΡΧΊΒψά¥Ω¥, Ι«ΦήΜΖκΡΒΡΜ·―ßΚœ≥…Ψ≠άζΝΥΕΰΗωΫΉΕΈ. ‘γΤΎΙ«ΦήΜΖκΡΒΡΚœ≥… «“‘≤ύΝ¥ΙΌΡήΆ≈»Ϊ±ΘΜΛΒΡœΏ–‘κΡΈΣΒΉΈο, ΆΖΈ≤Ά®ΙΐΥθΚœ ‘ΦΝΕχΜΖΜ·. ΗΟΖΫΖ®¥φ‘Ύ–μΕύ»±Βψ. άΐ»γ, –η“Σ‘Λ÷Τ≤ΩΖ÷Μρ»Ϊ≤Ω±ΘΜΛΒΡœΏ–‘ΕύκΡ; Ζ¥”ΠΈΜΒψ ÷–‘÷––Ρ≤ΜΡή±Θ≥÷ΚΆ»ί“ΉΖΔ…ζΕύΨέΜ·Η±Ζ¥”Π. …œ άΦΆΡ©±Ψ άΦΆ≥θ, ―–ΨΩ’ΏΖΔ’Ι≥ωΜ·―ß―Γ‘ώ–‘Ν§Ϋ”ΜΖΜ·Ζ¥”Π÷Τ±ΗΙ«ΦήΜΖκΡΒΡΖΫΖ®, ≥…ΙΠΫβΨωΝΥ¥ΪΆ≥ΥθΚœΖ®ΒΡ»±œί. Υδ»Μάζ Ζ…œ”–≤Μ…ΌΙΊ”ΎΜΖκΡΖΫΟφΒΡΉέ ω[12ΓΪ17], ΒΪ «Εύ ΐ «ΝΐΆ≥Ϋι…ήΙ«ΦήΜΖκΡΓΔ≤ύΝ¥ΜΖκΡΚΆΜΖκΡάύΥΤΈοΒΡΫχ’Ι, Ή®Ο≈Ϋι…ήΙ«ΦήΜΖκΡΒΡΚœ≥…ΒΡΉέ ωΈΡ’¬»‘Κή…Ό. Μυ”ΎΙζΦ …œΉνΫϋ”Ωœ÷–μΕύΩΈΧβΉιάϊ”Ο–¬–ΆΒΡΕύκΡΝ§Ϋ”Ζ¥”Π÷Τ±ΗΙ«ΦήΜΖκΡ, Έ“Ο«ΫΪΉ≈÷Ί¥”Μ·―ßΚœ≥…Ϋ«Ε»Ήέ ωΙ«ΦήΜΖκΡΒΡΉν–¬Ϋχ’Ι.
1 ΙΧœύΜΖΜ·≤Ώ¬‘
ΙΧœύΜΖΜ·÷ΗΕύκΡΆΖΈ≤≥…θΘΑΖΦϋΖ¥”Π‘Ύ ς÷§…œΆξ≥…. ΗΟ≤Ώ¬‘”–ΝΫΗωΟςœ‘ΒΡ”≈Βψ, »γΫΒΒΆΖ÷Ή”Φδ≈ωΉ≤ΦΗ¬ , Φθ…ΌΖ÷Ή”ΦδΕύΨέΜ·Η±Ζ¥”Π; Φθ…ΌΚœ≥…÷–ΒΡΖ÷άκΚΆ¥ΩΜ·ΒΡ¥Έ ΐ. “ΜΑψά¥ΥΒ, ΕύκΡΩ…≤…”ΟΝΫ÷÷ΖΫ ΫΙΧΕ®”Ύ ς÷§…œ, Φ¥άϊ”Ο≤ύΝ¥ΙΌΡήΜυΆ≈ΟΣΕ® ς÷§ΚΆάϊ”Ο C ΕΥτ»ΜυΙΧΕ®”Ύ ς÷§. «Α’Ώ‘ΎΙΧœύ…œΆξ≥…ΕύκΡ–ρΝ–ΒΡΤ¥Ϋ”Κσ, Ά―»Ξ N ΕΥΚΆC ΕΥ±ΘΜΛΜυ”Έάκ≥ωΉ‘”…Α±ΜυΚΆτ»Μυ, ‘Ό‘ΎΙΧœύ…œΆξ≥… CΕΥτ»ΜυΚΆN ΕΥΑ±ΜυΒΡΥθΚœΜΖΜ·. Κσ’Ώ‘ράϊ”Ο÷ςΝ¥ CΕΥτ»ΜυΟΣΕ® ς÷§, ‘ΎΕύκΡ–ρΝ–Τ¥Ϋ”Άξ≥…Κσ, ΜνΜ·ΕύκΡΒΡ C ΕΥ, ≤ΔΥφ÷°”κN ΕΥΉ‘”…Α±ΜυΖΔ…ζΖ÷Ή”ΡΎΑΖΫβΜΖΚœΖ¥”Π. ΒΎΕΰ÷÷ΖΫΖ®ΜΖΜ·ΚΆΕύκΡΒΡΙΧœύ«–Ην «Ά§ ±Ϋχ––ΒΡ, «“≤Μ–η“Σ≤ύΝ¥ΙΧΕ®, “ρΕχ±»ΒΎ“Μ÷÷ΖΫΖ®ΗϋΨΏ”≈ Τ. –η“Σ÷Η≥ω, ΙΧœύΜΖΜ·Ζ¥”Π≤ΜΡή±Θ÷ΛΖ¥”ΠΈΜΒψΒΡΙΙ–ΆΆξ»Ϊ±Θ≥÷, Ε‘”Ύ“Ήœϊ–ΐΒΡΑ±ΜυΥαΨΏ”–«±‘ΎΒΡΈΘœ’.
1.1 ≤ύΝ¥ΙΧΕ® ς÷§
άϊ”ΟΑ±ΜυΥα≤ύΝ¥ΙΌΡήΆ≈ΙΧΕ® ς÷§, œΏ–‘ΕύκΡ N ΕΥΑ±ΜυΚΆ C ΕΥτ»ΜυΆ®ΙΐΥθΚœΖ¥”Π…ζ≥…ΜΖκΡ «ΙΧœύΚœ≥…Ι«ΦήΜΖκΡΒΡ≥Θ”ΟΖΫΖ®. ΗΟΖΫΖ®–η“Σ’ΐΫΜΒΡ±ΘΜΛΜυ”Ο”Ύ±ΘΜΛΕύκΡ≤ύΝ¥ΙΠΡήΆ≈ΓΔN ΕΥΑ±ΜυΚΆ C ΕΥτ»Μυ. ≤ύΝ¥ΙΠΡήΆ≈“ΜΑψ≤…”ΟΕ‘ΥαΟτΗ–ΒΡ±ΘΜΛΜυ; N ΕΥΑ±ΜυΩ…≤…”Ο Fmoc; C ΕΥτ»Μυ≥Θ”ΟΕ‘ Pd ΟτΗ–ΒΡœ©±ϊθΞΫαΙΙ. ΨΏΧεΒΡ≤ΌΉς»γΆΦ 1 Υυ Ψ. Ήœ», ΒΎ“ΜΗωΑ±ΜυΥαάϊ”Ο≤ύΝ¥ΙΌΡήΆ≈ΚΆ ς÷§…œΙΌΡήΆ≈Ζ¥”ΠΙΧΕ®”Ύ ς÷§; ΗΟΑ±ΜυΥαΒΡΠΝ-Α±Μυ“‘Fmoc±ΘΜΛ, C-ΕΥτ»Μυ≤…”Οœ©±ϊθΞ±ΘΜΛ. Ϋ”œ¬ά¥, Α¥’’¥ΪΆ≥ Fmoc ΙΧœύΚœ≥…Άξ≥…ΡΩ±ξ–ρΝ–ΒΡΤ¥Ϋ”Κσ, Ζ÷±π”Ο Pd(PPh3)4ΓΔΏΏύΛ ‘ΦΝΫβ≥ΐ C ΕΥœ©±ϊθΞΚΆ N ΕΥ Fmoc ±ΘΜΛΜυ. ΉνΚσ, œΏ–‘ΕύκΡ ΉΈ≤Ά®ΙΐΥθΚœΖ¥”Π Βœ÷ΜΖΜ·, ≤Δ‘Ύ TFA Ής”Οœ¬”κ ς÷§Ζ÷άκ, Ήν÷’ΒΟΒΫΧλ»ΜΫαΙΙΒΡΜΖκΡ. ≥Θ”Ο≤ύΝ¥ΙΧΕ® ς÷§ΒΡΑ±ΜυΥαΈΣ Asp, Glu[18], His[19]Μρ’Ώ Lys[20]. 2011 Ρξ, Veerman Β»[21]άϊ”Ο Glu ≤ύΝ¥ΟΣΕ® ς÷§, Κœ≥…≥ωΜΖΉ¥»ΥΧεΆΌ“Κ÷–ΒΡΉιΑΖΥΊ, ≤Δ÷ΛΟςΤδΜν–‘ «Χλ»ΜœΏ–‘ΉιΑΖΥΊΒΡ1000 ±Ε. –η÷Η≥ω, ΗΟ≤Ώ¬‘‘ΎΙΧœύΚœ≥…÷–≥Θ≥ΘΜα”–ΕΰΆΣΏΏύΚΗ±Ζ¥”Π, «“ΜΖΜ·Ζ¥”Π≤ΜΡή»Ζ±ΘΖ¥”Π÷––ΡΒΡ ÷–‘±Θ≥÷.
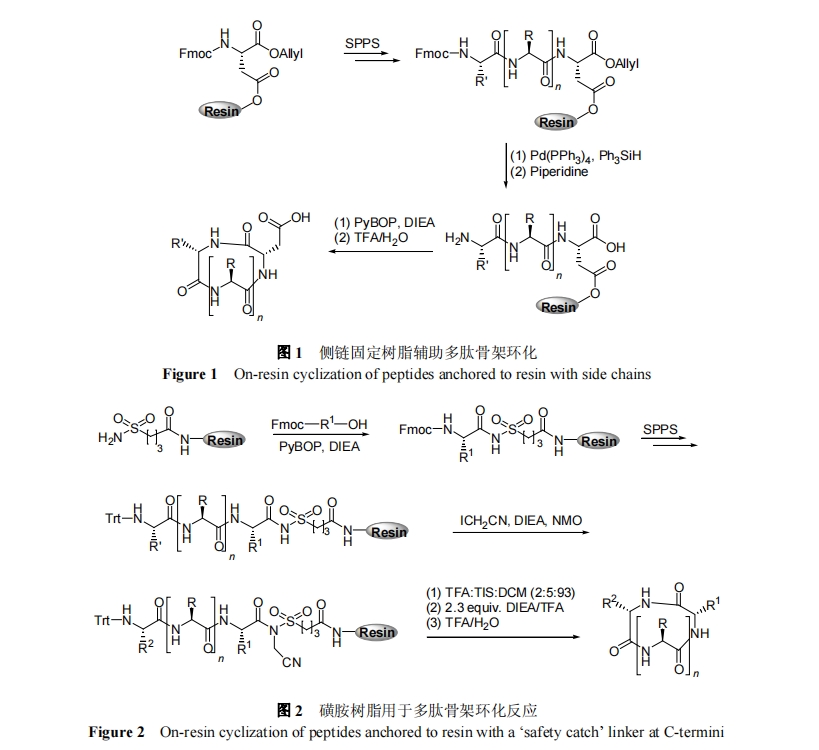
1.2 C ΕΥΜ«θΘΑΖΙΧΕ® ς÷§
‘Ύ Fmoc Ζ®ΙΧœύΕύκΡΚœ≥…÷–, ΕύκΡ C ΕΥτ»Μυ≤ΜΡή÷±Ϋ”≤…”ΟΜνΜ·θΞΜρΜνΜ·θΘΑΖΒΡ–Έ Ϋά¥ΙΧΕ® ς÷§. ’β «“ρΈΣFmoc ΕύκΡΙΧœύΚœ≥…≤ΌΉςΙΐ≥Χ–η“Σ Ι”Ο«ΩΦν–‘«ΉΚΥ–‘ ‘ΦΝΓΣΓΣΏΏύΛ. ‘Ύ≤…”Ο C ΕΥΙΧΕ® ς÷§ΖΫ ΫΚœ≥…Ι«ΦήΜΖκΡΙΐ≥Χ÷–, ΕύκΡ C ΕΥΝ§Ϋ”±έ±Ί–ηΕ‘ΏΏύΛΈ»Ε®, «“‘ΎœΏ–‘κΡΙΧœύΤ¥Ϋ”ΚσΩ…±ΜΗΏ–ßΉΣΜ·ΈΣΜνΜ·―θθΞΜρΜνΜ·θΘΑΖ, “‘ Βœ÷”κ N ΕΥΉ‘”…Α±ΜυΖΔ…ζΖ÷Ή”ΡΎΜΖΜ·Ζ¥”Π. Μ«θΘΑΖΫαΙΙ¬ζΉψΗΟ“Σ«σ, Ω…“‘”Ο”Ύ C ΕΥΟΣΕ® ς÷§ΙΧœύΚœ≥…Ι«ΦήΜΖκΡ. Μ«θΘΑΖΕ‘ΏΏύΛ ‘ΦΝΖ«≥ΘΈ»Ε®, ΕχΨ≠ΙΐΆιΜυΜ·ΚσΡήΉΣ±δΈΣΜνΜ·θΘΑΖ, Ω…“‘”κ¬ψ¬ΕΉ‘”…Α±ΜυΖΔ…ζΗΏ–ßΒΡΑΖΫβΖ¥”Π. ΨΏΧε≤ΌΉς»γΆΦ 2 Υυ Ψ. Ήœ»‘ΎΆξ≥…ΕύκΡΙΧœύΤ¥Ϋ”ΚσΫΪ N ΕΥΑ±Μυ±ΘΜΛΜυFmocΉΣΜ·ΈΣΕ‘ΥαΟτΗ–ΒΡTrt±ΘΜΛΜυ, Ϋ”œ¬ά¥Ζ÷±π”ΟΒβΜ·““κφ¥ΠάμΜ«θΘΑΖ ς÷§ΚΆ»θΥαΫβ≥ΐ N ΕΥ Trt ±ΘΜΛΜυ“‘ΦΑ”Ο”–ΜζΦν¥ΏΜ· ΉΈ≤ΙΧœύΜΖΚœΖ¥”Π, ΉνΚσάϊ”Ο«ΩΥα TFA Ϋβ≥ΐ≤ύΝ¥±ΘΜΛΜυΒΟΒΫΧλ»ΜΫαΙΙΒΡΙ«ΦήΜΖκΡ. 1999Ρξ, Morriello Β»[22]‘Ύ Merck ΙΪΥΨ Ή¥Έάϊ”Ο KennerΓ·s Μ«θΘΑΖ ς÷§≥…ΙΠΚœ≥…ΝΥΙ«ΦήΜΖκΡΚΆΜΖτ»Ζ”ΥακΡ. 2005 Ρξ, Ganesan Β»[23]≤…”ΟΗΟΖ®Κœ≥…ΝΥΚΘ―σΧλ»Μ≤ζΈοΙ«ΦήΜΖκΡkahalalide A ΦΑάύΥΤΈο, ≤ΔœΒΆ≥―–ΨΩΝΥΗΟΧλ»Μ≤ζΈοΒΡΫαΙΙΙΠΡήΙΊœΒ. ¥ΥΆβ, Μ«θΘΑΖ ς÷§ΜΙ”Ο”ΎΕύ’≥ΨζΥΊ[24]ΓΔΝ¥―τΟΙΥΊΒ»[25,26]ΜΖκΡΒΡΜ·―ßΚœ≥….
1.3 C ΕΥΖΦΜυθΘΜυκ¬ΙΧΕ® ς÷§
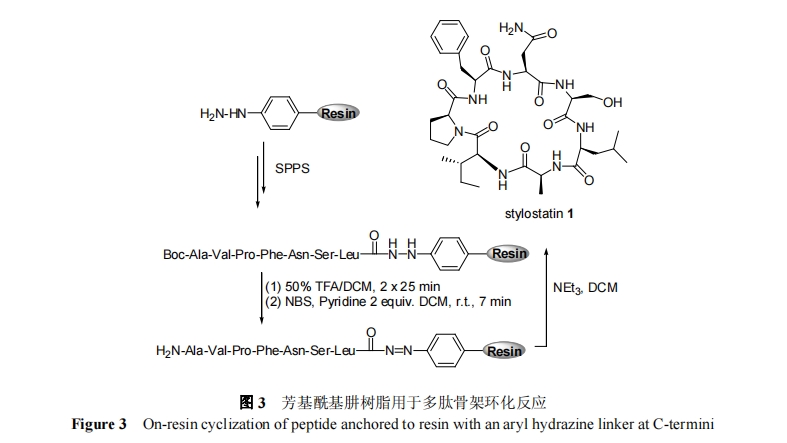
ΖΦΜυθΘΜυκ¬Ω…“‘Ά®ΙΐΆ―«β―θΜ·ΉΣΜ·ΈΣΜνΜ·θΘΑΖ, Α±ΜυΩ…“‘”κΜνΜ·θΘΑΖΖΔ…ζ≥…θΘΑΖΦϋΖ¥”Π. ―θΜ·Ιΐ≥ΧΕ‘Εύ ΐΑ±ΜυΥα≤Μ‘λ≥…Η±Ζ¥”Π, “ρΕχ‘≠άμ…œΩ…”Ο”ΎΕύκΡΒΡ C ΕΥΙΧœύΜνΜ·÷Τ±ΗΙ«ΦήΜΖκΡ. ΗΟΖ¥”Π‘γ‘Ύ 1970 Ρξ±Μ Birr Β»[27]ΖΔœ÷, ΒΪ «≥ΛΤΎ“‘ά¥≤ΔΟΜ”–±ΜΕύκΡ―–ΨΩ’ΏΥυΙΊΉΔ. ÷±ΒΫ1997 Ρξ, Langner Β»[28] Ή¥Έάϊ”Ο C ΕΥΖΦΜυθΘΜυκ¬Κœ≥…ΝΥΙ«ΦήΜΖΤΏκΡ, ≤Δ÷ΛΟςΗΟΖΫΖ®÷Τ±ΗΙ«ΦήΜΖκΡΒΡ–ß¬ ±»¥ΪΆ≥“ΚœύΥθΚœΗΏ. 2001 Ρξ, Waldmann Β»[29]άϊ”ΟΗΟΖ®“≤≥…ΙΠ÷Τ±ΗΝΥΙ«ΦήΜΖΝυκΡ, stylostatin, »γΆΦ 3 Υυ Ψ. ¥ΥΆβ, Janda Β»[30]≤ΔΫΪΗΟΖΫΖ®”Ο”ΎΕύ÷÷Ι«ΦήΜΖκΡΖ÷Ή”ΒΡΙΙΫ®. ÷ΒΒΟ“ΜΧα, 2004 Ρξ, Camareo Β»[31]άϊ”ΟΖΦΜυκ¬―θΜ·ΜνΜ·Ζ¥”ΠΖΔ’ΙΝΥ Fmoc ΝρθΞΒΡΚœ≥…ΖΫΖ®. Ά®ΙΐΫαΚœΉ‘»ΜΜ·―ßΝ§Ϋ”Ζ¥”Π, Camareo Β»[32]ΉνΫϋ Βœ÷ΝΥΖά”υΥΊΒΡΜ·―ßΚœ≥…. C ΕΥΖΦΜυθΘΜυκ¬ΜνΜ·ΜΖΚœ“‘…Χ“ΒΜ·ΒΡ±Ϋκ¬ ς÷§ΈΣ‘ΊΧεΫχ––ΙΧœύΚœ≥…, Άξ≥…ΕύκΡΤ¥Ϋ”Κσ, άϊ”Ο NBS ΜώΆ≠ ‘ΦΝ―θΜ·ΖΦΜυθΘΜυκ¬ Βœ÷ N ΕΥΑ±Μυ”κ C ΕΥΒΡΙΧœύΜΖΜ·Ζ¥”Π. –ηΉΔ“β, ΗΟΖΫΖ®Ε‘ΈΜΉη±»ΫœΟτΗ–, Κœ”ΎΖ¥”ΠΈΜΒψΈΜΉη±»ΫœΒΆΒΡΑ±ΜυΥαΒΡΜΖΜ·, «“―θΜ·ΧθΦΰΩ…Ρή‘λ≥… Met ΚΆ Trp ≤ύΝ¥ΒΡ―θΜ·Η±Ζ¥”ΠΖΔ…ζ.
2 “ΚœύΜΖΜ·≤Ώ¬‘
ΙΧœύΜΖΜ·≤Ώ¬‘≤Μ ΚœΝ¥±»Ϋœ≥ΛΒΡΙ«ΦήΜΖκΡΚœ≥…. ’β «”…”ΎΥφΉ≈κΡΝ¥ΒΡ‘ω≥Λ, ΙΧœύΖ¥”ΠΒΡ–ß¬ Μαœ‘÷χΫΒΒΆ. “ρ¥Υ, Ε‘”Ύ≥ΛΝ¥ΒΡΙ«ΦήΜΖκΡΗΏ–ßΒΡΚœ≥…ΖΫΖ®»‘ΈΣ“ΚœύΥθΚœ. ¥ΪΆ≥ΒΡ“ΚœύΥθΚœ «“‘≤ύΝ¥»Ϊ±ΘΜΛΒΡœΏ–‘κΡΈΣ‘≠Νœ, Ά®ΙΐΥθΚœΖ¥”ΠΕχΆξ≥…ΕύκΡΒΡΆΖΈ≤ΜΖΚœ. ≤ύΝ¥»Ϊ±ΘΜΛκΡΒΡ»ήΫβ–‘ ή–ρΝ–ΚΆΝ¥≥ΛΕ»”Αœλ¥σ, “ρ¥ΥΥθΚœ«Α–ηΕ‘”–Μζ»ήΦΝΫχ––”≈Μ·[16,17]. ΈΣΫΒΒΆΖ÷Ή”ΦδΥθΚœΗ±Ζ¥”Π, “ΚœύΥθΚœΒΡ≈®Ε»“ΜΑψ“Σ«σ―œΗώΩΊ÷Τ. Εχ«“, ¥ΪΆ≥ΥθΚœΖ¥”ΠΈΜΒψΝΔΧε±Θ≥÷–‘≤ν, “ΜΑψ ”Ο”Ύ C ΕΥΈΣ Gly ΚΆ Pro ΒΡΙ«ΦήΜΖΚœΖ¥”Π[33]. ΥφΉ≈κΡΝ¥Ε‘Ϋ”ΦΦ θΒΡΖΔ’Ι, Μ·―ß―Γ‘ώ–‘Ζ¥”Π≥…ΈΣΙ«ΦήΜΖκΡΚœ≥…ΒΡ–¬–ΥΖΫΖ®. œύ±»¥ΪΆ≥ΥθΚœΖ¥”Π, Μ·―ß―Γ‘ώ–‘Ζ¥”ΠΝΔΧε±Θ≥÷–‘ΗΏ, Ζ¥”ΠΧθΦΰΗϋΈΣΈ¬ΚΆΚΆΗΏ–ß, “ρΕχ≥…ΈΣΗΟΖΫΟφΫϋΤΎ―–ΨΩΒΡ÷ςΝς.
2.1 ΙΙœσΥχΕ®ΫαΙΙΗ®÷ζΙ«ΦήΜΖΜ·Ζ¥”Π
œΏ–‘ΕύκΡ‘Ύ»ή“Κ÷–“ΜΑψΟΜ”–ΫαΙΙ, ΒΦ÷¬Τδ C ΕΥΚΆ NΕΥ≈ωΉ≤ΒΡΦΗ¬ ≤ΜΗΏ, “ρΕχΖ÷Ή”ΡΎΜΖΜ·Ζ¥”ΠΥΌ¬ œύΕ‘Ϋœ¬ΐ. ΈΣΝΥΧαΗΏΖ÷Ή”ΡΎΙ«ΦήΜΖΜ·Ζ¥”ΠΒΡ–ß¬ , ―–ΨΩ’ΏΩ…“‘ΫΪœΏ–‘ΕύκΡΒΡΙΙœσœό÷Τ”ΎΜΖΜ·“ΉΖΔ…ζΒΡΙΙœσ. œό÷ΤœΏ–‘ΕύκΡΙΙœσΒΡΖΫΖ®“ΜΑψ”–ΝΫ÷÷, Φ¥ΡΎ≤Ω“ΐ»κΩ…ΗΡ‘λΕύκΡΫαΙΙΒΡ±ΘΜΛΜυΜρΆβΦ”Ω…¥ΌΫχΧΊΕ®ΙΙœσ–Έ≥…ΒΡΫπ τάκΉ”.
ΊΆΗ§Α±ΥαΫαΙΙΒΞ‘Σ“ΐ»κΕύκΡ–ρΝ–Ω…¥οΒΫœό÷ΤœΏ–‘ΕύκΡΙΙœσ¥ΌΫχΖ÷Ή”ΡΎΆΖΈ≤ΜΖΜ·Ζ¥”ΠΒΡ–ßΙϊ. ±ϊ≤φ±ΘΜΛΒΡSer, Thr Μρ Cys «≥ΘΦϊΒΡ»ΐ÷÷ΊΆΗ§Α±Υα, «“ΨυΩ…Ά®ΙΐΥα–‘ΧθΦΰΆ―»Ξ±ΘΜΛΜυΒΟΒΫΧλ»ΜΫαΙΙΒΡΑ±ΜυΥα. 1999 Ρξ, Mutter Β»[34]ΫΪΊΆΗ§Α±ΥαΉςΈΣ Thr ΒΡ«ΑΧε≥…ΙΠ Βœ÷ΝΥΙ«ΦήΜΖ»ΐκΡΒΡΚœ≥…, ≤Δ÷ΛΟςΜΖΜ·ΒΉΈοœΏ–‘ΕύκΡΒΡ≈®Ε»Ω…ΗΏ¥ο0.1 mol/L. 2004 Ρξ, Turner Β»[35]“ΐ»κ 3 Ε‘±ϊ≤φ±ΘΜΛ ThrΗ®÷ζΖ÷Ή”ΡΎΜΖΜ·, Βœ÷Ι«ΦήΜΖΝυκΡ[(Val-Thr)3]ΒΡΚœ≥…, »γΆΦ 4 Υυ Ψ. 2010 Ρξ, Jolliffe Β»[36]±®ΒάΝΥΗΟΖΫΖ®‘ΎΙ«ΦήΜΖΥΡκΡΚœ≥…ΖΫΟφΒΡ”Π”Ο.
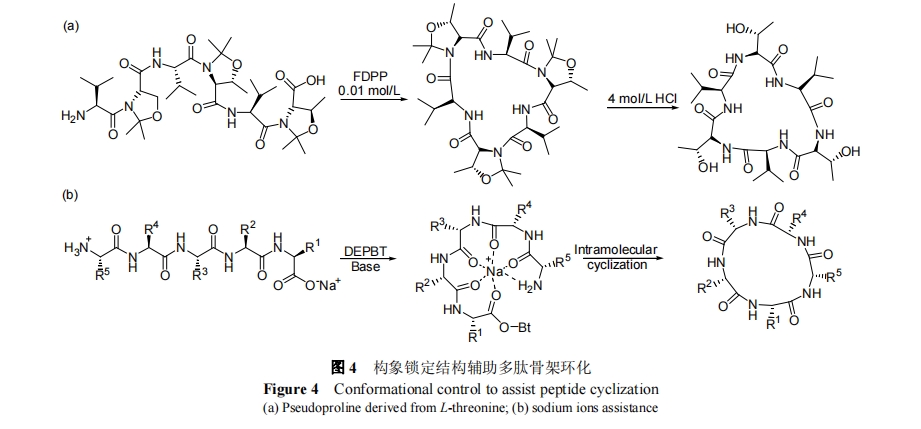
Ϋπ τάκΉ”Ά®Ιΐ”κΕύκΡ–ρΝ–÷–ΒΡΜυΆ≈≈δΈΜΉς”ΟΩ…“‘¥ΌΫχœΏ–‘ΕύκΡΆΖΈ≤Ω’ΦδΫ”Ϋϋ¥”Εχάϊ”ΎΖ÷Ή”ΡΎΜΖΜ·Ζ¥”ΠΒΡΖΔ…ζ. 2003 Ρξ, Ye Β»[37]ΖΔœ÷ NaΘΪΩ…“‘”κΕύκΡ÷– 5 ΗωΝΎΫϋΒΡτ ΜυΖΔ…ζ¬γΚœ¥ΌΫχΙ«ΦήΜΖΈεκΡΒΡΚœ≥…, »γΆΦ4Υυ Ψ. ΗΟ―–ΨΩΉι[38]‘Ύ 2005 ΡξΖΔœ÷ CsΘΪ“≤ΡήΆ®ΙΐάύΥΤΒΡΜζ÷Τ¥ΌΫχΙ«ΦήΜΖΤΏκΡΒΡΚœ≥….
2.2 Sanger ‘ΦΝΓΔMukaiyama ‘ΦΝΜρ’Ώ N ΕΥ»±ΒγΉ”
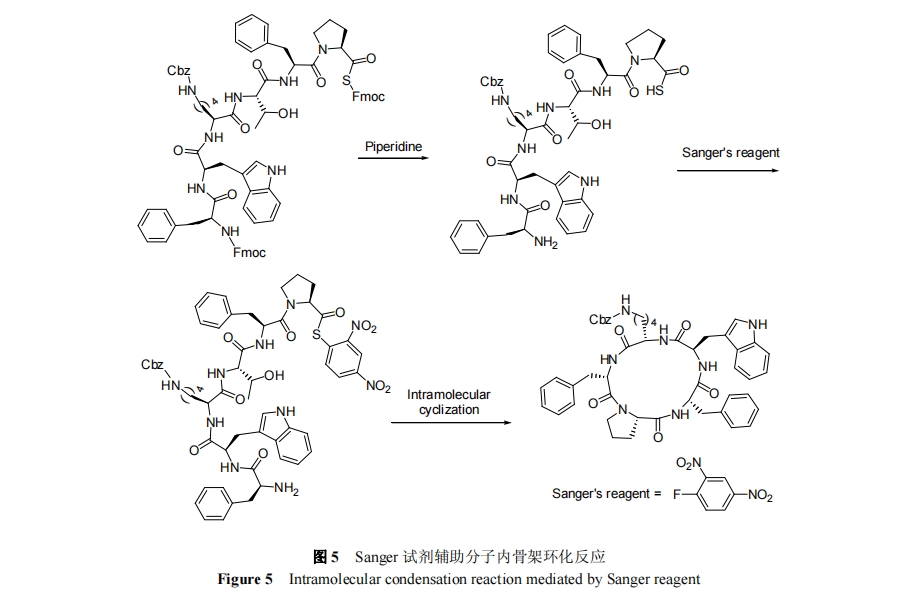
ΜυΆ≈»Γ¥ζΒΡΜ«ΑΖ ‘ΦΝΜνΜ· C ΕΥέœΜυΥακΡΜΖΜ·Ζ¥”ΠC ΕΥέœΜυΥακΡΩ…“‘”κ Sanger ‘ΦΝΓΔMukaiyama ‘ΦΝΜρ’Ώ N ΕΥ»±ΒγΉ”ΜυΆ≈»Γ¥ζΒΡΜ«ΑΖ ‘ΦΝΖ¥”ΠΉΣ±δΈΣΜν–‘ΫœΗΏΒΡ C ΕΥΖΦΜυΝρθΞκΡ. N ΕΥΉ‘”…ΒΡΑ±ΜυΩ…“‘”κΖΦΜυΝρθΞκΡΖΔ…ζΖ÷Ή”ΡΎΜΖΜ·Ζ¥”Π…ζ≥…Ι«ΦήΜΖκΡ. 2010 Ρξ, CrichΒ»[39ΓΪ41]≤…”Ο Sanger ‘ΦΝΜνΜ· C ΕΥέœΜυΥακΡ≥…ΙΠΚœ≥…ΝΥΙ«ΦήΜΖΈεκΡΚΆΙ«ΦήΜΖΝυκΡ, »γΆΦ 5 Υυ Ψ. ≤ύΝ¥ΙΌΡήΆ≈“ρ”ΑœλΗΟΖ¥”ΠΙΐ≥Χ“ΜΑψ≤…”ΟΥαΟτΗ–ΜυΆ≈Ϋχ––±ΘΜΛ, ‘ΎΜΖΜ·Ζ¥”ΠΆξ≥…Κσ, ±ΘΜΛΜυΩ…“‘‘ΎΥα–‘œ¬Ϋβ±ΘΜΛΒΟΒΫΧλ»ΜΫαΙΙΒΡΙ«ΦήΜΖκΡ.
2.3 Ϋπ τάκΉ”Μρ”–ΜζΦν¥ΏΜ·ΝρθΞκΡΙ«ΦήΜΖΜ·Ζ¥”Π
‘γ‘Ύ…œΗω άΦΆ 80 Ρξ¥ζ, »’±ΨΩΤ―ßΦ“ Aimoto Β»[42]ΖΔœ÷, ‘ΎΫπ τάκΉ” AgΘΪΒΡ¥ΏΜ·œ¬, ΕύκΡΝρθΞΩ…“‘”κΕύκΡΒΡΑ±ΜυΖΔ…ζΗΏ–ßΒΡΝ§Ϋ”Ζ¥”Π. «ΉΝρ–‘Ϋπ τάκΉ” AgΘΪΆ®Ιΐ”κC ΕΥΝρθΞ≈δΈΜΦ”«ΩΝΥΝρθΞΒΡάκ»Ξ–‘, ¥”Εχ¥ΌΫχΉ‘”…Α±Μυ”κΤδΖΔ…ζΑΖΫβΖ¥”Π.
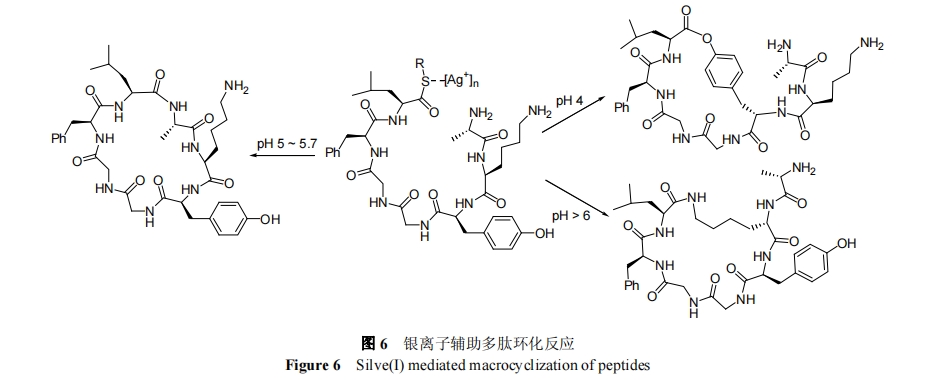
1999 Ρξ, Tam Β»[43]≤…”Ο AgΘΪ¥ΏΜ·ΕύκΡ N ΕΥΑ±Μυ”κ CΕΥΝρθΞΒΡΖ÷Ή”ΡΎΜΖΜ·Ζ¥”Π , Άξ≥…ΝΥΙ«ΦήΜΖΤΏκΡ[AIa-Lys-Tyr-GIy-GIy-Phe-Leu]ΒΡΜ·―ßΚœ≥…. ”–»ΛΒΡ «, Tam ―–ΨΩ±μΟς, Ά®ΙΐΒςΫΎ»ή“ΚΒΡΥαΕ», AgΘΪ¥ΏΜ·ΝρθΞΩ…―Γ‘ώ–‘”κ N ΕΥΑ±ΜυΓΔLys ≤ύΝ¥Α±ΜυΜρ Tyr ≤ύΝ¥Ζ”τ«ΜυΖΔ…ζΖ¥”Π, »γΆΦ 6 Υυ Ψ. ‘Ύ pH 4.0 ΒΡ»ή“Κ÷–, Lys ≤ύΝ¥Α±ΜυΚΆ N ΕΥΑ±ΜυΨυ±Μ÷ Ή”Μ·Εχ ß»ΞΖ¥”ΠΜν–‘, Tyr ≤ύΝ¥Ζ”τ«Μυ”≈œ»”κ C ΕΥΝρθΞΖΔ…ζΖ÷Ή”ΡΎΜΖΜ·…ζ≥…¥σΜΖΡΎθΞκΡ; ‘ΎpH 5.0ΓΪ6.0 ±, N ΕΥΑ±ΜυΚΆ C ΕΥΝρθΞ”≈œ»ΖΔ…ζΜΖΜ·Ζ¥”Π, …ζ≥…¥ΪΆ≥ΒΡΙ«ΦήΜΖΕύκΡ; ‘Ύ pHΘΨ6 ±, Lys ≤ύΝ¥Α±ΜυΜν–‘ΉνΗΏ, Ήνάϊ”ΎΖΔ…ζΖ÷Ή”ΡΎΜΖΜ·Ζ¥”Π≤ζ…ζ≤ύΝ¥ΜΖκΡΫαΙΙ. –η÷Η≥ω, AgΘΪ¥ΏΜ·ΝρθΞΒΡΙΐ≥Χ≤Δ≤ΜΉήΡή±Θ≥÷Ζ¥”ΠΈΜΒψ ÷–‘÷––ΡΒΡΙΙ–Ά, ”»ΤδΕ‘”ΎΖ¥”ΠΥΌ¬ ΫœΈΣΜΚ¬ΐΒΡΜΖΜ·Ιΐ≥Χ.
‘ΎΏδΏρΒΡ¥ΏΜ·œ¬, ΝρθΞ“≤Ρή±ΜΜνΜ·Εχ”κΑ±ΜυΖΔ…ζΖ÷Ή”ΡΎΜΖΜ·Ζ¥”Π. 2009 Ρξ, Houghten Β»[44]άϊ”ΟΚ§”–ΏδΏρΒΡ““κφ»ή“Κ(MeCNΓΟ1.5 mol/L ΏδΏρ)≥…ΙΠΚœ≥…ΝΥΙ«ΦήΜΖΈεκΡ. ΉνΫϋ, Houghten Β»[45]”÷ΖΔœ÷, ‘ΎΏδΏρ¥ΏΜ·œ¬ΥΩΑ±Υα≤ύΝ¥τ«Μυ“≤Ρή”κΝρθΞΖΔ…ζΖ÷Ή”ΡΎθΞΜ·Ζ¥”Π, Ω…”Ο”ΎΜ·―ßΚœ≥…Χλ»Μ≤ζΈο¥σΜΖΡΎθΞκΡ.
2.4 ΈόΚέ ©Χ’ΕΓΗώΙ«ΦήΜΖΜ·Ζ¥”Π
2000 Ρξ, Raines[46]ΚΆ Bertozzi Β»[47]œύΦΧΖΔ’Ι≥ωΈόΚέ ©Χ’ΕΓΗώΝ§Ϋ”Ζ¥”Π. ΗΟΖ¥”Π « ©Χ’ΕΓΗώΖ¥”ΠΒΡΗΡΫχ. ΨΏΧεΜζ÷ΤΈΣ, Ήœ»Κ§”–ΜζΝΉΚΆΝρθΞ–ό ΈΒΡΕύκΡΜα”κΒΰΒΣ–ό ΈΝμ“ΜΖ÷Ή”ΕύκΡΖ¥”Π…ζ≥…ΕύκΡΝΉ“ΕΝΔΒ¬÷–Φδ≤ζΈο, Ϋ”œ¬ά¥“ΕΝΔΒ¬÷–ΒΡ«ΉΚΥ–‘ΑΖΆ®ΙΐΖ÷Ή”ΡΎNΒΫS«®“Τ–Έ≥…θΘΑΖΦϋ, ΉνΚσΝΉΜυΚΆέœΜυ–ό ΈΜυΆ≈‘ΎΥ°÷–ΖΔ…ζΥ°ΫβΖ¥”ΠΕχάκ»ΞΆξ≥…ΕύκΡΤ§ΕΈΒΡΈόΚέΝ§Ϋ”.
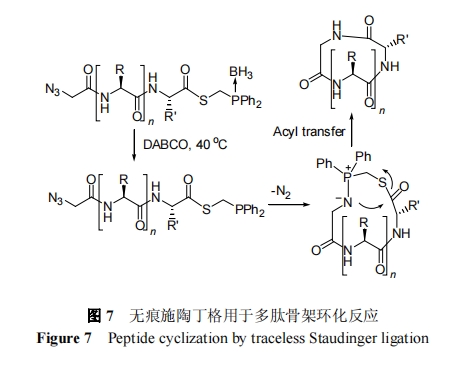
2008 Ρξ, Hackenberger Β»[48]ΫΪΈόΚέ ©Χ’ΕΓΗώΝ§Ϋ”Ζ¥”Π”Ο”ΎΖ÷Ή”ΡΎΖ¥”Π, Άξ≥…ΝΥ»ΐ÷÷Ι«ΦήΜΖ °“ΜκΡΒΡΜ·―ßΚœ≥…. ΗΟ―–ΨΩ±μΟςΈόΚέ ©Χ’ΕΓΗώΜΖΜ·Ζ¥”ΠΦ»Ω…”Ο”Ύ≤ύΝ¥±ΘΜΛκΡΒΡΆΖΈ≤ΜΖΜ·”÷Ω…”Ο”Ύ≤ύΝ¥≤Μ±ΘΜΛΕύκΡΒΡΆΖΈ≤―Γ‘ώ–‘ΜΖΜ·. ≤ύΝ¥±ΘΜΛκΡΒΡΆΖΈ≤ΜΖΜ· «≤…”Ο≈π ‘ΦΝ±ΘΜΛ”–ΜζΝΉ, ”Ο DABCO (1,4-ΕΰΒΣ‘”ΕΰΜΖ[2.2.2]–ΝΆι)―Γ‘ώ–‘Ϋβ≥ΐ≈πΜυ±ΘΜΛΜυ, ΆΖ≈ C ΕΥΝΉΜυ–ό ΈΝρθΞκΡ”Ο”ΎΖ÷Ή”ΡΎΜΖΜ·, »γΆΦ 7 Υυ Ψ. Υα–‘ΧθΦΰΫβ≥ΐ≤ύΝ¥Υυ”–±ΘΜΛΜυΒΟΒΫΒΡCΕΥΈΣΝΉ–ό ΈΝρθΞΚΆNΕΥΈΣΒΰΒΣ–ό ΈκΡΩ…÷±Ϋ”Ψ≠ΙΐΖ÷Ή”ΡΎΜΖΜ·Ζ¥”Π≤ζ…ζΧλ»ΜΙ«ΦήΜΖκΡ. –η÷Η≥ω, ΈόΚέ ©Χ’ΕΓΗώΜΖΜ·Ν§Ϋ”¥φ‘ΎΦΗΗω»±Βψ. άΐ»γ, Ζ¥”ΠΒΉΈοΝΉ ‘ΦΝ“Ή”Ύ±Μ―θΤχ―θΜ·; Ζ¥”Π÷––ηΦ””–Μζ»ήΦΝ“‘“÷÷ΤΥ°ΫβΗ±Ζ¥”Π; ΜΖΜ·Ζ¥”ΠΈΜΒψ“ΜΑψΈΣΈΜΉηΫœΒΆΒΡ Gly.
2.5 »©Μυ―θθΞ”κ Ser/Thr Ι«ΦήΜΖΜ·Ζ¥”Π
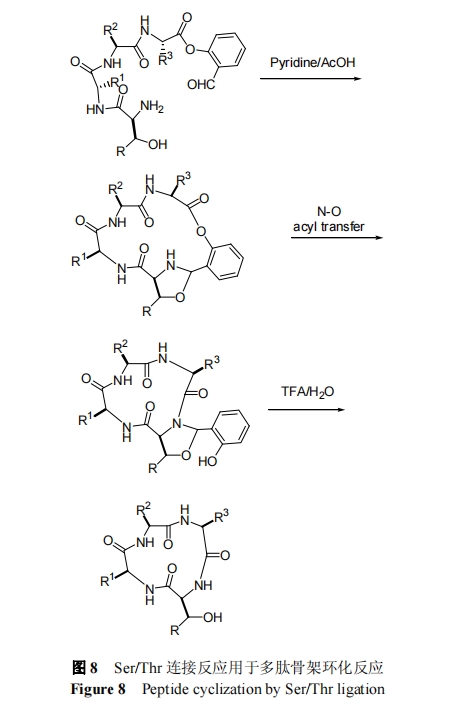
‘γ‘Ύ 1994 Ρξ, Tam Β»[49]ΖΔœ÷ C ΕΥΈΣ Π¬-τ«Μυ““»©–ό ΈΒΡ―θθΞκΡΩ…“‘―Γ‘ώ–‘”κ N ΕΥ Ser, Thr Μρ Cys ΖΔ…ζΜ·―ß―Γ‘ώ–‘≥…θΘΑΖΦϋΝ§Ϋ”Ζ¥”Π. ΈΣΫβΨω Tam ΖΫΖ®÷–Ζ¥”ΠΉΣΜ·ΥΌ¬ ΒΆΚΆ–ό ΈΜυΆ≈≤Μ“ΉΫβ≥ΐΒΡ»±Βψ, 2010 Ρξ, Li Β»[50]ΫΪΝΎτ«Μυ±ΫΦΉ»©Χφ¥ζ Π¬-τ«Μυ““»©, ΖΔ’Ι≥ω–¬“Μ¥ζ“‘ SerΚΆ Thr ΈΣΝ§Ϋ”ΈΜΒψΒΡΜ·―ß―Γ‘ώ–‘Ν§Ϋ”Ζ¥”Π, »γΆΦ 8 Υυ Ψ. œύ±»Εχ―‘, ΗΟΖ¥”Π–ß¬ œ‘÷χΧαΗΏ, ≤ΔΩ…”Ο”Ύ C ΕΥΈΣΗΏΈΜΉηΒΡ Val ΒΡΝ§Ϋ”. “ρΈΣΫαΙΙ÷–ΒΡΗ®÷ζΜυΆ≈Ω…“‘‘Ύ TFA ÷–ΗΏ–ß»Ξ≥ΐ, Υυ“‘ΗΟΖ¥”ΠΒΟΒΫΒΡ≤ζΈοΈΣΧλ»ΜΫαΙΙΒΡΙ«ΦήΜΖΕύκΡ. 2013 Ρξ, Li Β»[51,52]≤…”ΟΗΟΖΫΖ®Ζ÷±πΆξ≥…ΝΥ¥σΜΖΡΎθΞκΡ-¥οΆ–ΟΙΥΊΚΆΙ«ΦήΜΖΥΡκΡΒΡΚœ≥…. »ΜΕχ, ΗΟΖ¥”Π÷–Υυ–η C ΕΥ―θθΞκΡΒΡΚœ≥…≤ΌΉςœύΕ‘ΫœΈΣΗ¥‘”, «“Ζ¥”Π–η‘ΎΏΝύΛ““ΥαΒΡΜλΚœ»ήΦΝ÷–Ϋχ––[51].
2.6 ΆΣΥα-τ«ΑΖΜΖΜ·Ζ¥”Π
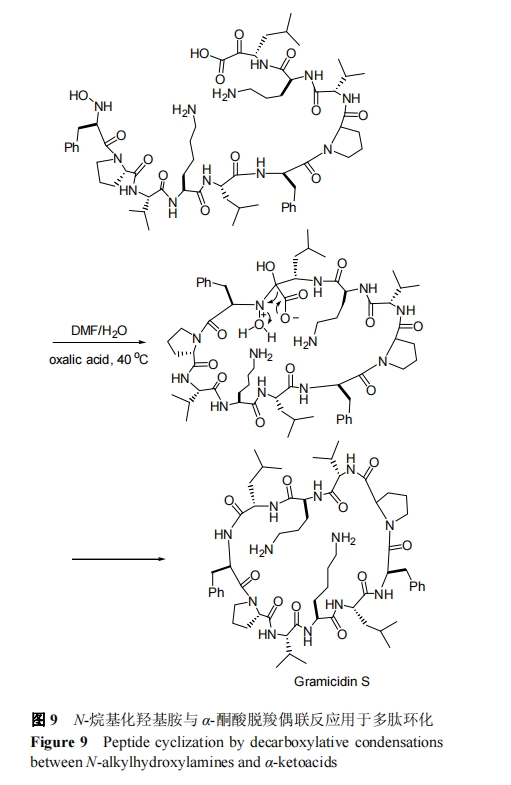
2006Ρξ, BodeΒ»[53]ΖΔœ÷CΕΥΆΣΥα–ό ΈΕύκΡΩ…“‘”κNΕΥτ«ΑΖΜ·–ό ΈΒΡΕύκΡΖΔ…ζΗΏ–ß―Γ‘ώ–‘≥…θΘΑΖΦϋΖ¥”Π. ΗΟΖ¥”ΠΒΡΜυ±Ψ‘≠άμ «τ«ΑΖ”κΆΣΥαΆ®ΙΐΖ÷Ή”ΡΎΆ―τ»ΚΆΆ―Υ°Ζ¥”Π–Έ≥…Χλ»ΜθΘΑΖΦϋ. 2012 Ρξ, Bode Β»[54]‘Υ”ΟΖ÷Ή”ΡΎΒΡΆΣΥα-τ«ΑΖΖ¥”Π≥…ΙΠΆξ≥…ΝΥΜΖ 10 κΡ-ΕΧΗΥΨζκΡΒΡΜ·―ßΚœ ≥…, »γΆΦ 9. –η÷Η≥ω, ΗΟΖ¥”Π“ΜΑψ“Σ«σ‘ΎΜΙ”– DMF ΒΡ”–Μζ»ήΦΝ÷–Ϋχ––, «“Ζ¥”ΠΈ¬Ε»‘Ύ 40 ΓφΉσ”“ ±Ζ¥”ΠΥΌ¬ ΫœΈΣΗΏ–ßΓΘ
3 Ή‘»ΜΜ·―ßΝ§Ϋ”≤Ώ¬‘
1994 Ρξ, Kent Β»[55ΓΪ61]ΖΔ’ΙΒΡΉ‘»ΜΜ·―ßΝ§Ϋ”Ζ¥”Π «”Π”ΟΉνΈΣΙψΖΚΒΡΒΑΑΉ÷ ΒΡΜ·―ßΚœ≥…ΖΫΖ®. Φ¥‘Ύ pH ΈΣ 7ΚΆΝρ¥Φ¥φ‘Ύœ¬, “ΜΖ÷Ή” CΕΥΈΣΝρθΞΒΡκΡΝ¥”κΝμ“ΜΧθN ΕΥΈΣΑκκΉΑ±ΥαΒΡκΡΩ…“‘ΖΔ…ζΗΏ–ßΒΡ≥…θΘΑΖΦϋΝ§Ϋ”Ζ¥”Π. Ή‘»ΜΜ·―ßΝ§Ϋ”Ζ¥”ΠΨ≠ΙΐΜ·―ß―Γ‘ώ–‘≤ΕΜώΚΆΖ÷Ή”ΡΎ÷Ί≈≈ΝΫΗω≤Ϋ÷ηΕχΆξ≥…. Ήœ», ΝΫΖ÷Ή”ΒΡ C ΕΥΝρθΞΚΆ N ΕΥ Cys ≤ύΝ¥έœΜυ÷°ΦδΖΔ…ζΩ…ΡφΒΡΝρ¥Φ-ΝρθΞΫΜΜΜ…ζ≥…“ΜΖ÷Ή”ΒΡΝρθΞ÷–ΦδΧε, Τδ¥Έ, Ά®ΙΐΖ÷Ή”ΡΎ≤ΜΩ…ΡφΒΡ SΓΣN θΘΜυ«®“Τ, …ζ≥…Χλ»ΜκΡΦϋ. Ή‘»ΜΜ·―ßΝ§Ϋ”Ζ¥”ΠΒΡ”≈Βψ «: ΗΏ–ßΓΔΗΏ―Γ‘ώ–‘ΓΔ≤ύΝ¥Άξ»ΪΈό–η±ΘΜΛ. Ι«ΦήΜΖκΡΩ…“‘Ά®ΙΐΖ÷Ή”ΡΎΒΡΉ‘»ΜΜ·―ßΝ§Ϋ”Ζ¥”Π÷Τ±Η, Φ¥“ΜΧθΆ§ ±Κ§”– C ΕΥΝρθΞΚΆN ΕΥ Cys ΒΡœΏ–‘κΡΩ…“‘Ά®ΙΐΖ÷Ή”ΡΎΒΡ≥…θΘΑΖΦϋΖ¥”Π Βœ÷ΆΖΈ≤Ν§Ϋ”, »γΆΦ 10 Υυ Ψ. 1997 Ρξ, Tam Β»[62] Ή¥Έ”ΟΖ÷Ή”ΡΎΉ‘»ΜΜ·―ßΝ§Ϋ”Ζ¥”ΠΚœ≥…ΝΥ÷≤ΈοΜΖΕύκΡ Cyclotide. ΥφΚσ, –μΕύ―–ΨΩ’ΏΨυ≤…”ΟΉ‘»ΜΜ·―ßΝ§Ϋ”Ζ¥”ΠΆξ≥…ΝΥΙ«ΦήΜΖκΡΒΡΚœ≥…. ‘≠‘ρ…œ, Ή‘»ΜΜ·―ßΝ§Ϋ”ΜΖΚœ≤Ώ¬‘ τ”Ύ“ΚœύΜΖΜ·Ζ¥”ΠΒΡ“Μ÷÷. Μυ”ΎΗΟΖΫΟφΒΡ―–ΨΩΫœΕύ, Έ“Ο«Ε‘ΤδΉ®Ο≈ΒΡΫι…ή.
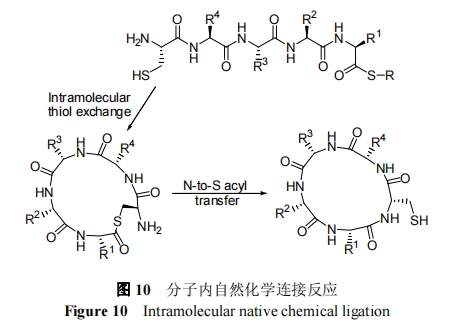
3.1 Ζ÷Ή”ΡΎέœΜυά≠Ν¥ ΫΉ‘»ΜΜ·―ßΙ«ΦήΜΖΜ·Ζ¥”Π
Ζ÷Ή”ΡΎέœΜυά≠Ν¥Ι«ΦήΜΖΜ·Ζ¥”Π «÷ΗΗΜΚ§ΑκκΉΑ±ΥαΒΡκΡΆ®ΙΐΖ÷Ή”ΡΎέœΜυ-ΝρθΞΫΜΜΜΖ¥”Π, ¥ΌΫχΕύκΡ C ΕΥΝρθΞ”κ N ΕΥ Cys Ω’ΦδΫ”ΫϋΕχΆξ≥…Ζ÷Ή”ΡΎΉ‘»ΜΜ·―ßΝ§Ϋ”Ζ¥”Π, »γΆΦ 11. ’β÷÷Ζ¥”ΠΖΫΖ®–η“Σ 3 ÷÷÷Ί“ΣΙΌΡήΆ≈: N ΕΥCysΓΔC ΕΥΝρθΞΚΆΡΎ≤ΩΉ‘”…έœΜυ. Ήœ», άκ C ΕΥΝρθΞΫϋΒΡΑκκΉΑ±Υα≤ύΝ¥«ΉΚΥ–‘ΫχΙΞΝρθΞ, Φ¥Νρ¥Φ-ΝρθΞΫΜΜΜ–Έ≥…Ζ÷Ή”ΡΎΝρθΞ; Τδ¥Έ, Ά®ΙΐΖ÷Ή”ΡΎΩ…ΡφΒΡΉΣΝρθΞΜ·Ιΐ≥Χ, –Έ≥…Ηϋ¥σΒΡΖ÷Ή”ΡΎΝρθΞ, ΙΒΟ C ΕΥΚΆ N ΕΥΫχ“Μ≤ΫΫ”Ϋϋ; Ήν÷’, Ά®Ιΐ≤ΜΩ…ΡφΒΡ SΓΣN θΘΜυΉΣ“ΤΖ¥”Π–Έ≥…ΡΎθΘΑΖ. 1999 Ρξ, Tam Β»[63] Ή¥ΈΧα≥ωΝΥΖ÷Ή”ΡΎέœΜυά≠Ν¥ΜΖΜ·Ζ¥”ΠΜζάμ, ≤Δ÷Η≥ωά≠Ν¥ΜΖΜ·Ζ¥”Π «λΊ«ΐΕ·ΒΡΙΐ≥Χ, Φ¥Ά®Ιΐ–ΓΒΡάκ…Δ÷–Φδ≤ζΈο ΙΒΟΖ¥”Π±»œύ”ΠΒΡ“Μ≤ΫΜΖΚœΖ¥”ΠΗϋΗΏ–ß. ΨΓΙή―–ΨΩ÷ΛΟςΖ÷Ή”ΡΎ≤ΩΉ‘”…Νρ¥ΦΩ…“‘Φ”ΩλΖ÷Ή”ΡΎΒΡΉ‘»ΜΜ·―ßΖ¥”ΠΥΌ¬ , ΒΪ «≤Δ≤ΜΡή»ΖΕ®ΉΣΝρθΞΜ·“ΜΕ®―ΊΉ≈Ν¥œύΦΧΒΊΖΔ…ζ, Ά§ ±“≤≤ΜΡή»ΖΕ®Υυ”–ΒΡΉ‘”…Νρ¥ΦΕΦ≤Έ”κ¥ΏΜ·Ιΐ≥Χ[64].
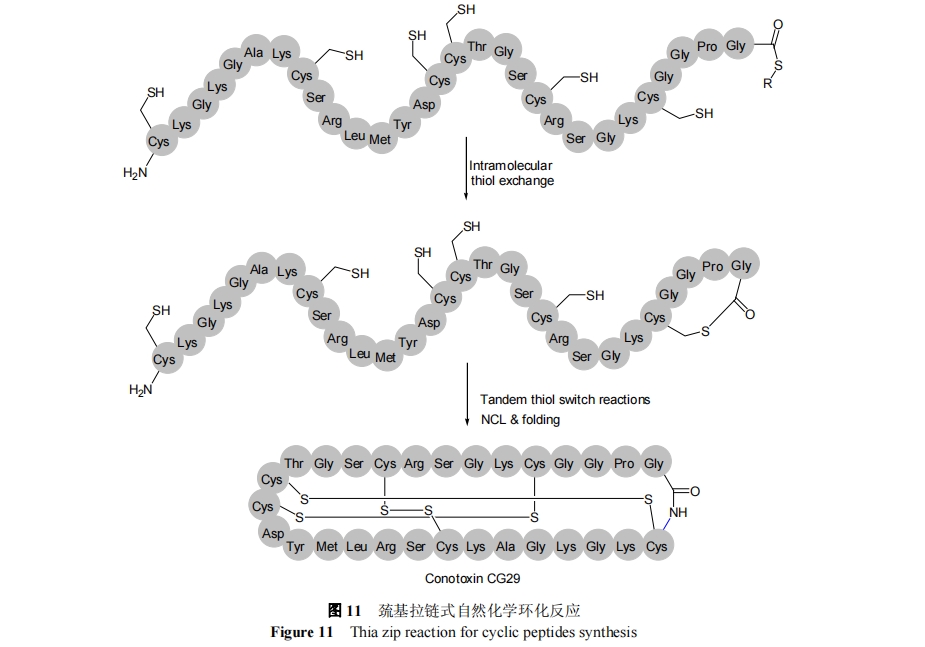
3.2 C ΕΥέœΜυ–ό ΈΖΦΜυ―θθΞκΡ≤Έ”κΉ‘»ΜΜ·―ßΙ«ΦήΜΖΜ·Ζ¥”Π
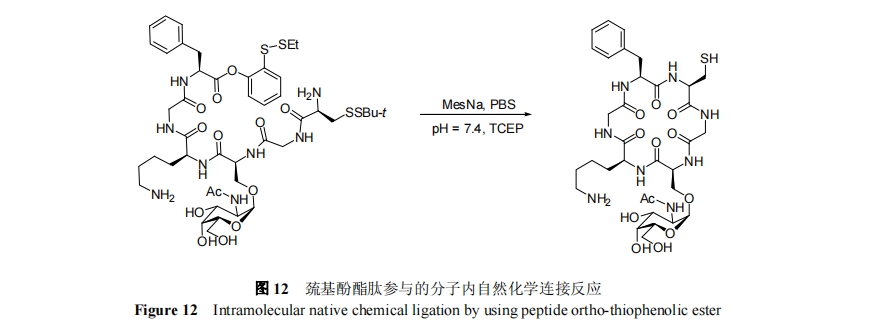
2004Ρξ, DanishefskyΒ»[65]ΖΔœ÷CΕΥΚ§”–ΝΎέœΜυ–ό ΈΒΡΖΦΜυ―θθΞκΡΩ…“‘Ά®ΙΐOΒΫSΖ÷Ή”ΡΎθΘΜυ«®“Τ‘≠ΈΜ…ζ≥…ΝρθΞκΡ, Βœ÷”κ N ΕΥ Cys κΡΒΡΜ·―ß―Γ‘ώ–‘Ν§Ϋ”Ζ¥”Π. άϊ”ΟΖΦΜυ―θθΞκΡΉςΈΣΝρθΞκΡΒΡ«ΑΧεΚΆΖ÷Ή”ΡΎΉ‘»ΜΜ·―ßΝ§Ϋ”Ζ¥”Π, Danishefsky Β»[66]”Ύ 2006 Ρξ Βœ÷ΝΥΚ§”–ΒΞΧ«–ό ΈΒΡΙ«ΦήΜΖΝυκΡΒΡΜ·―ßΚœ≥…, »γΆΦ 12 Υυ Ψ.
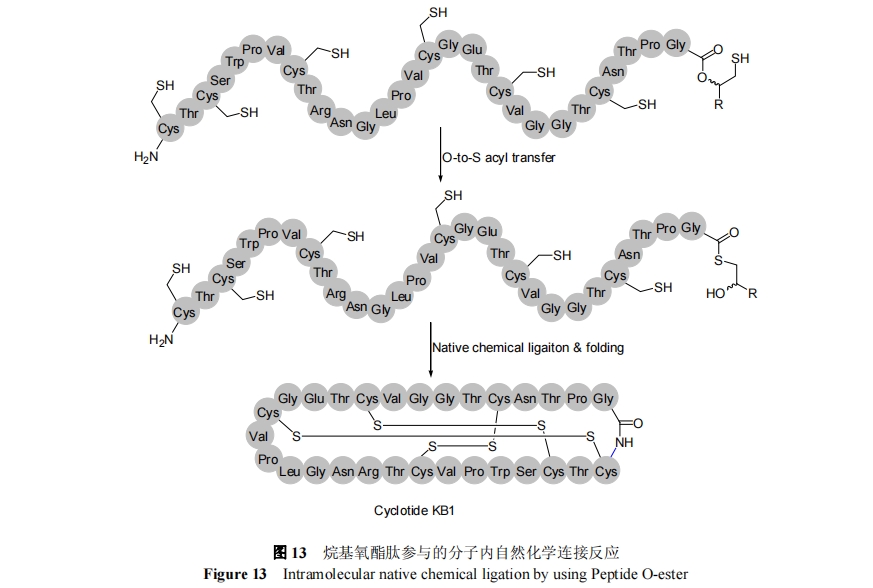
3.3 C ΕΥέœΜυ–ό ΈΆιΜυ―θθΞκΡ≤Έ”κΉ‘»ΜΜ·―ßΙ«ΦήΜΖΜ·Ζ¥”Π
ΝρθΞΦϋΕ‘ΦνΚΆ«ΉΚΥ ‘ΦΝΖ«≥Θ≤ΜΈ»Ε®, “Ή”ΎΚΆΏΏύΛΖΔ…ζΖ¥”Π, “ρΕχ, ≤ΜΡή÷±Ϋ”≤…”ΟFmocΖ®ΙΧœύΚœ≥…, Εχ÷ΜΡή≤…”Ο Boc Ζ®Ϋχ––÷Τ±Η. ¥ΪΆ≥ΒΡ Boc-SPPS ÷Τ±ΗΝρθΞ÷––η“Σ Ι”ΟΦΪΈΣΈΘœ’ΒΡ HF, «“¥χ”–Εύ÷÷Ζ≠“κΚσ–ό ΈκΡΒΡΕ‘HF Ψυ≤ΜΈ»Ε®, »γΧ«–ό ΈΚΆΝΉΥαΜ·–ό ΈΒ». ΈΣΫβΨω’β“ΜΈ Χβ, ΫϋΡξά¥“ΣΖΔ’ΙΝΥΕύ÷÷”––ßΒΡ Fmoc-SPPS Κœ≥…ΕύκΡΝρθΞΒΡΖΫΖ®. OΓΣS «®“ΤΝρθΞ÷Τ±ΗΖ® « Fmoc Ζ®÷Τ±ΗΝρθΞκΡΒΡ“Μ÷÷÷Ί“ΣΖΫΖ®. ΗΟΖΫΖ®άϊ”ΟΆιΜυ―θθΞΉςΈΣΝρθΞΒΡ«ΑΧε, Ά®ΙΐΖ÷Ή”ΡΎ OΓΣS «®“Τ‘≠ΈΜ–Έ≥…ΝρθΔϔΎΉ‘»ΜΜ·―ßΝ§Ϋ”Ζ¥”Π[67ΓΪ69]. 2012 Ρξ, Liu Β»[70]≤…”Ο―θθΞκΡ, Ά®ΙΐΖ÷Ή”ΡΎΒΡΉ‘»ΜΜ·―ßΝ§Ϋ”Ζ¥”ΠΆξ≥…ΝΥ÷≤ΈοΙ«ΦήΜΖΕύκΡ KB1ΒΡΚœ≥…, »γΆΦ 13 Υυ Ψ. ―θθΞ”κ Fmoc ΙΧœύΚœ≥…Φφ»ί, “ρΕχ÷Τ±ΗΤπά¥±»ΝρθΞΗϋΈΣΖΫ±ψ. »ΜΕχ–ηΉΔ“β, OΓΣS «®“ΤΫωΫω ”Ο”ΎΈΜΉηΫœ–ΓΒΡΑ±ΜυΥα, Ε‘”ΎΗΏΈΜΉηΒΡΜΖΜ·Ζ¥”Π, ¥φ‘ΎΥ°ΫβΗ±Ζ¥”Π.
3.4 C ΕΥέœΜυ–ό ΈθΘΑΖκΡ≤Έ”κΒΡΉ‘»ΜΜ·―ßΙ«ΦήΜΖΜ·Ζ¥”Π
”κ―θθΞκΡάύΥΤ, ―–ΨΩ’ΏΖΔœ÷Εύ÷÷ C ΕΥΚ§έœΜυ–ό ΈΒΡθΘΑΖκΡ“≤Ω…ΉςΈΣΝρθΞκΡΒΡ«ΑΧε. 2011 Ρξ, Macmillan Β»[71]“‘ C ΕΥΈΣΕΰκΡ Gly-CysΓΔN ΕΥΈΣ Cys ΒΡΧλ»ΜœΏ–‘ΕύκΡΈΣΒΉΈο, άϊ”ΟΕΰκΡ Gly-Cys ΒΡΖ÷Ή”ΡΎΒΡ N-S «®“ΤΝρθΞΜ·Μζ÷Τ, Άξ≥…ΝΥΕύ÷÷ΩΙΨζΙ«ΦήΜΖΕύκΡΒΡΚœ≥…. 2013 Ρξ, TamΒ»[72]“‘ C ΕΥΈΣ N-έœ““Μυ-N-ΆιΜυΜ·θΘΑΖΕύκΡΈΣΒΉΈο, άϊ”ΟN-ΆιΜυΜ·θΘΑΖ“Ή”ΎΖΔ…ζΖ÷Ή”ΡΎΒΡN-S«®“ΤΒΡΧΊ’ς, Άξ≥…ΝΥ÷≤ΈοΙ«ΦήΜΖΕύκΡ Kalata B1 ΒΡΚœ≥…. ”κ―θθΞœύΥΤ, CΕΥθΘΑΖκΡΕ‘”ΎΈΜΉηΫœΗΏΒΡΜΖΜ·Ζ¥”ΠΫœ≤ν.
3.5 C ΕΥθΘκ¬κΡ≤Έ”κΒΡΉ‘»ΜΜ·―ßΙ«ΦήΜΖΜ·Ζ¥”Π
Υδ»ΜΙζΦ …œ”–ΚήΕύΩΈΧβΉιΖΔ’ΙΝΥ“ΜœΒΝ–”––ßΒΡFmoc-SPPS ΦδΫ”Κœ≥…ΕύκΡΝρθΞΒΡΖΫΖ®, ΒΪ «Ψυ¥φ‘ΎΈ Χβ, »γ–η“Σ‘Λ÷ΤΫαΙΙΗ¥‘”ΒΡΝ§Ϋ”±έΓΔ≥…ΝρθΞΖ¥”Π ήΈΜΉη”Αœλ¥σΚΆΒΉΈοΖ¥”ΠΜν–‘≤Μ“ΥΩΊ÷ΤΒ». 2011 Ρξ, Liu Β»[73ΓΪ78]“‘θΘκ¬ΕύκΡΉςΈΣΒΉΈο, ΖΔ’Ι≥ωθΘκ¬Ν§Ϋ”ΦΦ θ, ΫβΨωΝΥ«Α»ΥΖΫΖ®ΒΡΈ Χβ, ≥…ΈΣΕύκΡΦΑΒΑΑΉΚœ≥…ΖΫΟφΒΡ“ΜΗω÷Ί“Σ≥…Ιϊ.
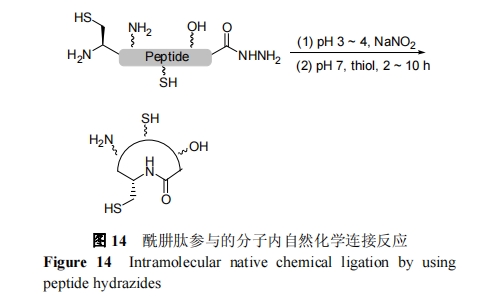
θΘκ¬Ν§Ϋ”ΦΦ θΒΡΜυ±Ψ‘≠άμ «, ‘Ύ»θΥαΧθΦΰœ¬, ΕύκΡθΘꬑΎ―«œθΥαΒΡ―θΜ·œ¬ΉΣΜ·ΈΣθΘΜυΒΰΒΣ, ΥφΚσ”κΝρ¥ΦΖ¥”Π‘≠ΈΜ…ζ≥…κΡΝρθΞ≤Δ÷±Ϋ””κNΕΥCysκΡΉ‘»ΜΜ·―ß―Γ‘ώ–‘Ν§Ϋ”Ζ¥”Π. ±Ψ÷ …œά¥ΥΒ, ’β÷÷Ν§Ϋ”ΖΫΖ® «θΘκ¬ΚΆΝρθΞ‘≠ΈΜΉΣ±δ, «“Μ÷÷ΗΡΫχΒΡΉ‘»ΜΜ·―ßΝ§Ϋ”Ζ¥”Π. ΕύκΡθΘꬓ≤Ω…“‘Ά®Ιΐ…ζΈο±μ¥οΫχ––÷Τ±Η. 2012 Ρξ, LiuΒ»[79]≤…”ΟΖ«Χλ»Μ―θθΞΑ±ΜυΥα«Ε»κΦΦ θ≥…ΙΠ±μ¥ο≥ωΕύκΡθΘκ¬, ΖΔ’ΙΝΥ±μ¥οΒΑΑΉθΘκ¬Ν§Ϋ”ΦΦ θ. Ά§Ρξ, Liu ΩΈΧβΉι[80]‘Υ”ΟΖ÷Ή”ΡΎθΘκ¬Ν§Ϋ”Ζ¥”Π, ≥…ΙΠ÷Τ±ΗΝΥœΒΝ–¥σ–ΓΒΡΙ«ΦήΜΖΕύκΡ, »γΆΦ 14 Υυ Ψ. ÷ΒΒΟΉΔ“β, Liu Β»[81]÷ΛΟςΖ÷Ή”ΡΎθΘκ¬Ζ¥”ΠΩ…“‘”Ο”ΎΗΏ–ß÷Τ±Η’≈ΝΠΫœ¥σΒΡΙ«ΦήΜΖΥΡκΡ. œύ±»«Α»ΥΖΫΖ®, ΕύκΡθΘκ¬ΨΏ”–Κœ≥…Ρ―Ε»ΒΆΓΔ≥…±ΨΒΆΝ°ΓΔΉ‘Ε·Μ·Φρ±ψΒΡ”≈ Τ. ¥ΥΆβ, θΘκ¬Ν§Ϋ”ΦΦ θΒΡΝ§Ϋ”–߬ ≤Μ ήΈΜΉη”Αœλ, Ε‘”ΎΨχ¥σΕύ ΐΈΜΒψΒΡΝ§Ϋ”Ψυ ”Ο.
4 «ΑΨΑΚΆ’ΙΆϊ
20 άΦΆ 40 Ρξ¥ζ, ΗΏΥΙΒ»ΖΔœ÷÷χΟϊΒΡ÷ΈΝΤ…ΥΩΎΗ–»ΨΒΡΕΧΗΥΨζκΡ, »Ο―–ΨΩ’ΏΩΣ ΦΙΊΉΔΙ«ΦήΜΖΕύκΡ―–ΨΩ. ‘ΎΙΐ»ΞΒΡΫΪΫϋΤΏ °Ρξάο, ΩΤ―ßΦ“‘ΎœΗΨζΓΔ’φΨζΓΔ÷≤ΈοΚΆΕ·Έο÷–ΨυΖΔœ÷Ι«ΦήΜΖκΡ, ≤Δ”–…Ό ΐΙ«ΦήΜΖκΡ“―≥…ΈΣ÷ΈΝΤ»ΥάύΦ≤≤ΓΒΡ“©ΈοΖ÷Ή”. ΉνΫϋ, ―–ΨΩ’ΏΖΔ’Ι≥ωΙΠΡή“©ΈοΕύκΡΓΑΦόΫ”Γ±Cyclotide ΦΦ θ, ΈΣΙ«ΦήΜΖκΡ“©Έο―–ΖΔΧαΙ©ΝΥ“ΜΧθ«Ω”–ΝΠΒΡΙΛΨΏ. 2012 Ρξ, Tam Β»[82]ΫΪ φΜΚΦΛκΡ B1 ήΧεόΉΩΙκΡΦόΫ”ΒΫ Cyclotide Ι«ΦήΡΎ, ΩΣΖΔ≥ω÷ΈΝΤΧέΆ¥ΒΡΩ…ΩΎΖΰΒΡΕύκΡ“©ΈοΖ÷Ή”. 2013 Ρξ, Camareo Β»[83]‘Υ”Ο…ζΈοΖΫΖ®ΫΪ…ζΈοΜν–‘ΕΧκΡPMIΦόΫ”ΒΫCyclotide, ≤Δ‘Ύ–Γ σΧεΡΎ»Ζ»œΗΟΜΖκΡΡήΙΜΆ®ΙΐΦΛΜν P53 ΒΑΑΉά¥“÷÷Τ÷ΉΝω…ζ≥Λ. ‘Ύ’βΦΗ °Ρξάο, Μ·―ß―–ΨΩ’Ώ“≤ΖΔ’ΙΝΥΕύ÷÷Ι«ΦήΜΖκΡΚœ≥…ΒΡΖΫΖ®. ¥”ΙΧœύΜΖΜ·Ζ¥”ΠΒΫ“ΚœύΜΖΜ·Ζ¥”Π, ¥”¥ΪΆ≥ΥθΚœΦΝΥθΚœΖ¥”ΠΒΫΫϋΤΎΖΔ’ΙΒΡΜ·―ß―Γ‘ώ–‘Ν§Ϋ”Ζ¥”Π, Μ·―ßΖΫΖ®“―Ψ≠÷πΫΞ≥…ΈΣ÷Τ±ΗΙ«ΦήΜΖκΡΒΡ÷Ί“Σ ÷ΕΈ. ΥφΉ≈Ι«ΦήΜΖκΡΚœ≥…ΦΦ θΒΡ≥… λΚΆΤδΫαΙΙ”κΙΠΡήΖΫΟφΒΡ…ν»κ―–ΨΩ, Έ“Ο«œύ–≈ΫΪ”–ΗϋΕύΒΡΙ«ΦήΜΖκΡ“©ΈοΖ÷Ή”±Μ»ΥΟ«ΖΔœ÷.
Οβ‘π…υΟςΘΚ±ΨΈΡΈΣ––“ΒΫΜΝς―ßœΑΘ§Αφ»®Ιι‘≠Ής’ΏΦΑ‘≠‘”÷ΨΥυ”–Θ§»γ”–«÷»®Θ§Ω…ΝΣœΒ…Ψ≥ΐΓΘΈΡ’¬±ξΉΔ”–Ής’ΏΦΑΈΡ’¬≥ω¥ΠΘ§»γ–η‘ΡΕΝ‘≠ΈΡΦΑ≤ΈΩΦΈΡœΉΘ§Ω…‘ΡΕΝ‘≠‘”÷ΨΓΘ
