
еЊвЊ: ЫцзХЖрыФРрЛЏКЯЮядкжЮСЦжзСіМВВЁКЭЩњЮявНбЇгІгУжаЕФживЊадгыШеОудіЃЌПЊЗЂКЭЙЙНЈЖрыФЗжзгЕФаТЗНЗЈвбГЩЮЊгаЛњКЯГЩЛЏбЇМвЕФбаОПШШЕуЁЃНќФъРДгаЛњЕчЛЏбЇзїЮЊвЛжжТЬЩЋИпаЇЕФЗДгІЙЄОпБЛж№ВНдЫгУгкгаЛњаЁЗжзгКЯГЩСьгђЃЌЦфЮТКЭПЩПиЕФЬиеїЪЪКЯНтОіЯжгаЩњЮяХМСЊВпТдДцдкЕФЛЏбЇКЭЧјгђбЁдёадЮЪЬтЃЌЮЊЖрыФЗжзгЕФбЁдёадаоЪЮЬсЙЉСЫвЛжжживЊКЯГЩЪжЖЮЁЃзлЪіСЫНќЮхФъРћгУЕчЛЏбЇЪжЖЮаоЪЮАБЛљЫсКЭЖрыФРрЛЏКЯЮяЕФЗДгІЃЌВћЪіСЫЕчЛЏбЇКЯГЩММЪѕЕФгХЪЦМАЦфдкПЊЗЂаТаЭЩњЮяЯрШнадЗНЗЈжаЕФЪЪгУадЁЃ
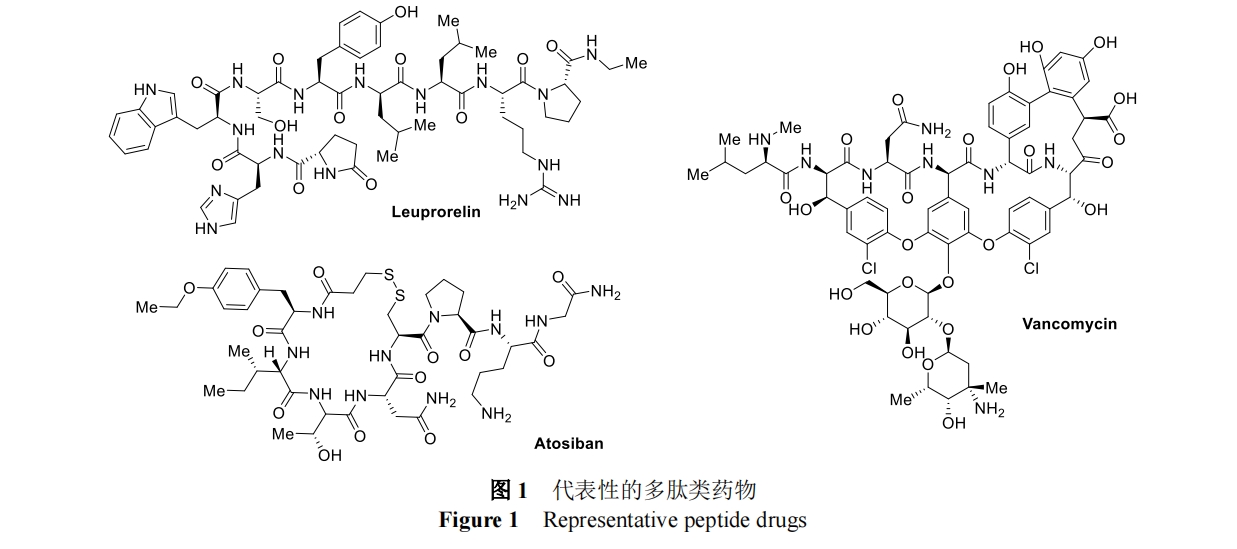
ЖрыФРрЛЏКЯЮяОпгаЛюадИпЁЂЬивьадЧПЁЂЖОИБзїгУаЁЁЂАаЕубЁдёадИпЕШгХЕуЃЌЖдАЉжЂЁЂздЩэУтвпадМВВЁЁЂМЧвфСІМѕЭЫЁЂОЋЩёЪЇГЃЁЂИпбЊбЙКЭФГаЉаФбЊЙмМАДњаЛМВВЁ[1]гаЯджјЕФСЦаЇКЭЙуЗКЕФгІгУЧАОАЁЃЫцзХШЋЧђаЁЗжзгвЉЮябаЗЂФбЖШЕФдіМгЃЌЛЏбЇаоЪЮыФвђЦфзПдНЕФЩњЮяЛюадКЭвЉДњЖЏСІбЇЬиадЪмЕНСЫПЦбЇМвУЧЕФЙуЗКЙизЂ[2]ЁЃФПЧАЃЌШЋЧђЪаГЁЩЯДѓдМга 80 жжЖрыФвЉЮяЃЌГЌЙ§ 150 жжЖрыФДІгкСйДВПЊЗЂНзЖЮЃЌСэга 400-600 жжЖрыФе§дкНјааСйДВЧАбаОП[3]ЃЌЦфжавдПЙжзСіЁЂПЙЩњЫигыУтвпЕїНкМСОгЖрЃЌШчПЙжзСівЉЮяССБћШ№Сж LeuprorelinЁЂПЙЩњЫивЉЮяЭђЙХУЙЫи Vancomycin КЭМЄЫиРрвЉЮяАЂЭаЮїАр AtosibanЃЈЭМ 1ЃЉЁЃЕЋАБЛљЫсКЭЖрыФРрЛЏКЯЮяЕФИпбЁдёадКЯГЩШдДцдкаэЖрРЇФбЃЌШчАБЛљЫсКЭЖрыФЗжзгЕФЮШЖЈадВюЁЂУєИаВрСДЃЈШчлЯЛљЃЉЕФДцдкЁЂБЃГжЪжаджааФЕФЭъећадЃЌвдМАШмНтадКЭДПЛЏЕШЮЪЬтЁЃ
ЫцзХгаЛњКЯГЩММЪѕЕФВЛЖЯЬсЩ§ЃЌНќФъРДАБЛљЫсКЭЖрыФЕФЛЏбЇбЁдёадКЯГЩЕУЕНСЫвЛЖЈЕФЗЂеЙ[4]ЁЃ2018 ФъЃЌTimothyПЮЬтзщБЈЕРСЫЙтДпЛЏаоЪЮАБЛљЫсЁЂЖрыФКЭЕААзжЪЕФбаОПНјеЙ[5]ЁЃдкЙтбѕЛЏЛЙдЙ§ГЬжаЃЌгаЛњЙтДпЛЏМСдкЮќЪеФмСПКѓдОЧЈЕНМЄЗЂЬЌЃЌдкИУзДЬЌЯТЃЌКмШнвзгызїЮЊЕчзгЙЉЬхЛђЪмЬхЕФЕзЮяЗЂЩњЕЅЕчзгзЊвЦЃЈSETЃЉЃЌаЮГЩздгЩЛљжаМфЬхЁЃгыЦфЫќЗНЗЈЯрБШЃЌЙтДпЛЏПЩдкЮТКЭЕФЬѕМўЯТЖдАБЛљЫсЛђЖрыФНјааЛЏбЇаоЪЮЃЌЭЌЪБРћгУПЩМћЙтзїЮЊПЩГжајФмдДЁЃЕЋЙтУєМСдкЫЎНщжЪжаЕФШмНтадВюЃЌДЫЭтЃЌДѓЖрЪ§вбБЈЕРЕФгУгкАБЛљЫсаоЪЮЕФЙтбѕЛЏЛЙдЗНЗЈашвЊЪЙгУЕБСПЕФЛЙдМСЃЈШчгаЛњМюЃЉЛђбѕЛЏМСЃЈШчИпМлН№ЪєбЮЃЉЁЃетаЉЪдМСЕФЪЙгУгыАБЛљЫсКЭЖрыФЕФЩњЮяаджЪВЛЯрШнЃЌДгЖјзшАСЫЙтДпЛЏЗНЗЈЕФгІгУЁЃC−H ЙйФмЛЏзїЮЊвЛжжЧПДѓЕФЙЄОпвбБЛЙуЗКгІгУгкАБЛљЫсКЭЖрыФЕФбЁдёадаоЪЮЃЌ2014 ФъЃЌBrimble ПЮЬтзщЖдАБЛљЫсКЭЖрыФКЯГЩжаЕФ C−H ЙйФмЛЏЗНЗЈНјааСЫзлЪі[6]ЁЃ2018 ФъЃЌAckermann ПЮЬтзщзмНсСЫЭЈЙ§ЮЛжУбЁдёад C−H ЛюЛЏЪЕЯжЖрыФКѓЦкЖрбљЛЏЕФбаОПНјеЙ[7]ЁЃ2020 ФъЃЌГТЙПЮЬтзщзлЪіСЫН№ЪєДпЛЏ CЈDH ЙйФмЛЏЃЈCHFЃЉЖдЖрыФНјаазщзАКѓаоЪЮЗНУцЕФбаОП[8]ЁЃЖрыФЕФЬивьадКЯГЩДгОжЯоЕФМЋадЧзКЫВаЛљЃЈШчАыызАБЫсКЭРЕАБЫсЃЉЃЌЭиеЙЕНЪшЫЎаджЌЗОзхКЭЗМЯузхВаЛљЁЃН№ЪєДпЛЏЕФ CHF ЗДгІЭЈЙ§ИФБфЗДгІЭООЖЁЂЪдМСКЭХфЬхРДПижЦЗДгІЛюадЃЌДгЖјЪЕЯжИќгаМјБ№адЕФЖрыФКЯГЩЁЃШЛЖј C−H ЙйФмЛЏЗНЗЈЭЈГЃашвЊЖдЛюадЮЛЕуНјааБЃЛЄЛђНшжњЕМЯђЛљЭХЃЌвдМѕЩйИБВњЮяЕФВњЩњЃЛЭЌЪБашвЊН№ЪєДпЛЏМСвдМАЛЏбЇМЦСПЕФЧПбѕЛЏМСЃЌЮоЫЎЮобѕЕФЗеЮЇЃЌетаЉЗДгІЬѕМўЯрЖдПСПЬЃЌЯожЦСЫИУЗНЗЈНјвЛВНЕФЭиеЙКЭгІгУЁЃ
ЕчЛЏбЇКЯГЩММЪѕзїЮЊвЛжжЮТКЭМАЛЗОГгбКУЕФКЯГЩЙЄОпЃЌНќФъРДЪмЕНСЫгаЛњКЯГЩЛЏбЇМвЕФЧрэљ[9]ЁЃДг 1848 Фъ KolbeбєМЋбѕЛЏЗДгІЃЌЕчЛЏбЇвбОЗЂеЙГЩвЛЯюживЊЕФММЪѕЪжЖЮЃЌЮЊЛЏбЇзЊЛЏЬсЙЉТЬЩЋПЩГжајЕФЕчзгЁЃШчЯЉЬўЕФЫЋЙйФмЛЏ[10]ЁЂСЂЬхбЁдёаддгЛЗКЯГЩ[11]ЁЂгы CO2 НјааєШЛЏЗДгІ[12]ЃЌЛђгІгУгкКЯГЩНсЙЙИДдгЕФЬьШЛВњЮя[13]ЁЃЕчЛЏбЇКЯГЩММЪѕЕФжївЊгХЪЦЪЧРћгУЕчзгЬцДњбѕЛЏЛЙдЪдМСЃЌЭЈЙ§ЕчМЋЕчЮЛЕФОЋШЗПижЦРДЕїПиЗДгІЕФШШСІбЇЧ§ЖЏСІЃЌЪЕЯжЮТКЭЬѕМўЯТЕФЛЏбЇзЊЛЏЃЌОпгаТЬЩЋЛЗБЃЕФЬиЕу[14]ЁЃЕЋЪЧЃЌдкЕчЛЏбЇЛЗОГжааоЪЮАБЛљЫсКЭЖрыФвРШЛДцдквЛаЉЬєеНЁЃдк Brabec КЭ MornsteinНЬЪкЕФдчЦкбаОПжа[15]ЃЌ20 жжГЃМћЕФЕААзжЪдадАБЛљЫсжЛгаАыызАБЫсЃЈ-0.22 VЃЉЁЂРвАБЫсЃЈ+0.93 VЃЉЁЂЩЋАБЫсЃЈ+1.02VЃЉЁЂзщАБЫсЃЈ+1.17 VЃЉКЭЕААБЫсЃЈ+1.48 VЃЉВрСДОпгабѕЛЏЛЙдЛюад[16]ЃЌПЩдкЕчМЋБэУцБЛбѕЛЏЃЌЫЕУїЖрыФжаДцдкЖржжЛюадЙйФмЭХПЩБЛЕчМЋбѕЛЏЛђЛЙдЁЃЖјЧвЃЌетаЉАБЛљЫсВрСДЕФбѕЛЏЛЙдЕчЪЦЛсЫцзХВЛЭЌЕФЛЗОГЃЌАќРЈ pH жЕвдМАгыжЎЯрСЌЕФыФСДЛђЕААзжЪЕФБфЛЏЖјЗЂЩњИФБфЁЃвђДЫЃЌЖдгкАБЛљЫсКЭЖрыФЕФЕчЛЏбЇаоЪЮашзЂвтЕчЮЛЗЖЮЇЃЌЪЕЯжЕчЮЛЕФОЋзМПижЦЃЌЗРжЙВрСДИБЗДгІЕФЗЂЩњЁЃ
2021 ФъЃЌMalins ПЮЬтзщвРОнЕчНтЗНЪНЖдЖрыФКЭЕААзжЪЕФЕчЛЏбЇаоЪЮЗНЗЈНјааСЫЙщФЩгызмНс[14]ЃЌЕЋЪЧНќФъРДЭЈЙ§ЕчЛЏбЇММЪѕЖдУєИаАБЛљЫсКЭЖрыФНјаабЁдёадаоЪЮЕФВпТдЕУЕНСЫПьЫйЗЂеЙЃЌаэЖраТгБЖРЬиЕФбаОПЯрМЬЖјГіЁЃБОЮФНсКЯЙњФкЭтПЮЬтзщНќЮхФъЕФБЈЕРЃЌЖдетвЛСьгђЕФЙЄзїНјааВЙГфгыЭъЩЦЃЌвРОнАБЛљЫсЕФжжРрМАЯрЙиЛЏбЇЗДгІРраЭЃЌЗжБ№ДгЃЈ1ЃЉАБЛљЫсВаЛљгыЖрыФВрСДЕФЕчЛЏбЇаоЪЮЃЛЃЈ2ЃЉМИжжГЃМћЗДгІРраЭЕФАБЛљЫсКЭЖрыФЕчЛЏбЇаоЪЮЃЛвдМАЃЈ3ЃЉЕчЛЏбЇЖрыФКЯГЩШ§ВПЗжФкШнНјааЙщФЩгызмНсЃЌЦкЭћЮЊНёКѓАБЛљЫсКЭЖрыФРрЛЏКЯЮяЕФЕчЛЏбЇаоЪЮЬсЙЉвЛЖЈЕФВЮПМзїгУЁЃ
1 АБЛљЫсВаЛљгыЖрыФВрСДЕФЕчЛЏбЇаоЪЮ
1.1 АБЛљЫсВаЛљЕФЕчЛЏбЇаоЪЮ
1.1.1 РвАБЫс
Gouin ПЮЬтзщдк 2018 ФъЪзДЮБЈЕРСЫ e-Y-click ЗДгІ[17]ЃЈЭМ 2AЃЉЁЃетжжЕчЛЏбЇЗНЗЈНЈСЂдк Barbas аЁзщ 2010 ФъЗЂБэЕФПЊДДадЙЄзїЩЯЃЌЦфУшЪіСЫРвАБЫсВрСДгыЛЏбЇбѕЛЏЩњГЩЕФШЁДњБНЛљ-3H-1,2,4-Ш§пђ-3,5(4H)-ЖўЭЊЃЈPTADsЃЉжЎМфЕФЗДгІ[18]ЁЃетжжЁАЕуЛїЪНЁБЗНЗЈБЛКЯГЩНчЙуЗКВЩгУЃЌЬсЙЉСЫЖржжРвАБЫсЙІФмЛЏЗжзгЃЌАќРЈПЙЬх-вЉЮяНсКЯЮя[19]ЁЂЗХЩфадБъМЧыФ[20]ЁЂЬЧНсКЯЮяКЭЬЧНсКЯвпУч[21]ЁЂDNA-ЕААзжЪНсКЯЮя[22]ЁЂвдМАЕААзжЪЫЎФ§НК[23]ЕШЁЃШчЩЯЫљЪіЃЌИУЗНЗЈвРРЕгкыхпђЧАЬхЕФДѓСПЛЏбЇбѕЛЏЗДгІвдЩњГЩЛюад PTADsЃЌЕЋетжждЄЛюЛЏВпТдЕФШБЕуЪЧдкЛЏбЇбѕЛЏМСДцдкЕФЧщПіЯТЃЌЛюЛЏЕФPTADs ЗжНтКЭСђДМбѕЛЏПЩФмЛсЖдРвАБЫсжЇСДдьГЩЁАЙ§ЖШаоЪЮЁБЁЃЮЊСЫОЁСПМѕЩйетаЉИБЗДгІЃЌашЖюЭтЪЙгУжњШмМСЛђЧхГ§МСЃЈШч TrisЃЉЁЃгыжЎЯрБШЃЌe-Y-click ЗДгІЭЈЙ§дкДПЫЎЛКГхвКжаНјааЮТКЭЕФЕчЛЏбЇбѕЛЏЃЈ0.36 VЃЌКубЙЕчНтЃЉРДШЁДњЁАЖлЛЏЁБЛЏбЇбѕЛЏМСЃЌДгЖјБмУтСЫетаЉЮЪЬтЁЃаЁЗжзгыхпђдкЕчМЋБэУцЕФСЌајдЮЛЕчЛЏбЇЛюЛЏЃЌЪЙРвАБЫсЙВщюЗДгІдкЗДгІЮяЩњГЩКѓСЂМДЗЂЩњЃЌвжжЦСЫВЛБивЊЕФИБЗДгІЃЌДгЖјЯджјЬсИпСЫбЁдёадЁЃДЫЭтЃЌМЋЕЭЕФгІгУЕчЮЛПЩзюДѓЯоЖШЕиМѕЩйбѕЛЏЫ№ЩЫвдМАгыДѓСПЕААзжЪАБЛљЫсВрСДЙйФмЭХЕФНЛВцЗДгІЁЃДгаЁыФМЄЫиДпВњЫиЕН 152kDa ЕФЕЅПЫТЁПЙЬхEpratuzumabЃЌетжжЮТКЭЕФЕчЛЏбЇЗНЗЈЭЈЙ§ЖдИїжжЕзЮяНјааЙІФмЛЏЃЌЬхЯжСЫЗЧГЃгХвьЕФЪЕгУадЁЃИќживЊЕФЪЧЃЌОЙ§e-Y-click аоЪЮКѓЃЌФПБъУИЦЯЬбЬЧбѕЛЏУИЕФЙІФмЛюадЕУвдБЃСєЁЃ
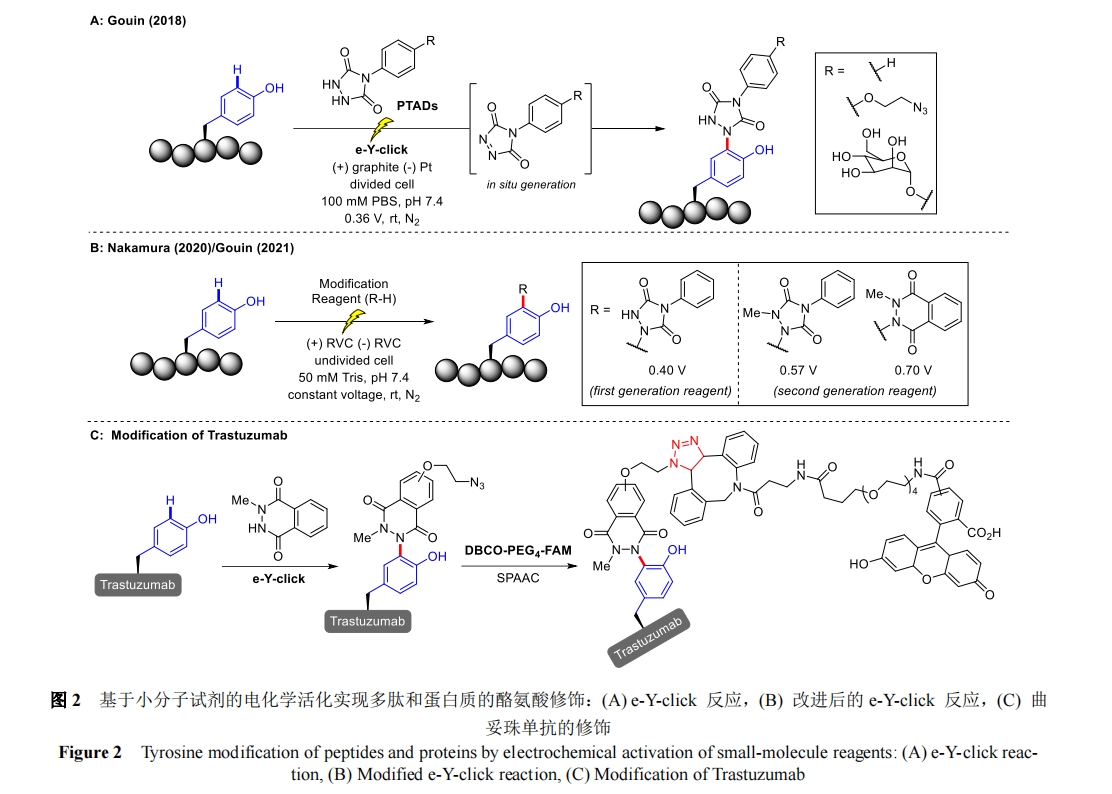
гЩгк e-Y-click ЗДгІВйзїМђБуЃЌОпгаСМКУЕФЩњЮяЯрШнадЃЌИУЗДгІКѓајБЛгУгкдкРвАБЫсВаЛљЩЯНЛСЊыФКЭЕААзжЪЕзЮяЃЌЮоашН№ЪєДпЛЏМСЛђЙтееМДПЩМьВтЦфШ§ЮЌНсЙЙ[24]ЁЃетЯюбаОПАќКЌСЫдк 0.36 V ЯТНјааЕФ e-Y-click ЗДгІвдМАдк-3.0 V ЯТЖдНЛСЊЪдМСжаДцдкЕФЖўСђМќНјааЕФЕчЛЏбЇЛЙдЁЃ
Nakamura ПЮЬтзщРћгУИФНјЕФ e-Y-click ЗДгІЃЌдкБэУцБЉТЖЕФРвАБЫсВаЛљЩЯНјааЮЛЕубЁдёадЩњЮяХМСЊЃЈЭМ 2BЃЉ[25]ЁЃЫћУЧЪЙгУ N-МзЛљТГУзХЕКЭ 1-МзЛљ-4-БНЛљыхпђбмЩњЮязїЮЊЕчЛЏбЇЛюадаЁЗжзгаоЪЮЪдМСЃЌГЩЙІЕидкЬьШЛРвАБЫсВаЛљЩЯаоЪЮСЫЪмБЃЛЄЕФАЫыФбЊЙмНєеХЫи IIЁЃгызюГѕЕФБНЛљыхпђЯрБШЃЌЕкЖўДњЪдМСЃЈN-МзЛљТГУзХЕКЭ 1-МзЛљ-4-БНЛљыхпђЃЉЕФЗДгІаЇТЪЖМгаЫљЬсИпЃЌGouin ПЮЬтзщдк 2021 ФъЕФвЛЯюбаОПжавВНјвЛВНжЄЪЕСЫетвЛЯжЯѓ[26]ЁЃЕЋЖдИќДѓЕФЕААзжЪШЫБэЦЄЩњГЄвђзгЪмЬх-2ЃЈHER2ЃЉПЙЬхЧњЭзжщЕЅПЙНјаааоЪЮЪБЃЌБНЛљыхпђКЭ N-МзЛљТГУзХЕОљНјеЙЫГРћЃЌЖј 1-МзЛљ-4-БНЛљыхпђЕФаоЪЮаЇЙћШДКмВюЃЌетвЛЪЕбщНсЙћЕФдвђЛЙашвЊНјвЛВНЬНОПЁЃДЫЭтЃЌИУЗДгІВЩгУШЋЫЎадЛКГхвКЃЈTrisЃЌ50 mMЃЉЁЂжаад pH жЕЃЈ7.4ЃЉКЭЮТКЭЕФЕчЛЏбЇЬѕМўЃЈ4-7 VЃЉЃЌЭЈЙ§ 2 ВНЙйФмЛЏЪЕЯжСЫЮЛЕубЁдёадЁЃЪзЯШЪЙгУКЌЕўЕЊЛЏЮяЕФаЁЗжзгЖдЧњЭзжщЕЅПЙНјаа e-Y-click аоЪЮЃЌШЛКѓдйгУЙйФмЛЏЖўБНВЂЛЗаСЯЉЃЈDBCOЃЉЪдМСНјааЮоЭЕуЛїЗДгІЃЈЭМ 2CЃЉЁЃетСНИіВНжшЪЧГЩЙІБъМЧЫљБиаыЕФЃЌВЂЧвЖдШмМСБЉТЖЮЛЕуЖМИќгаРћЃЌвђДЫгыЕЅВНЗНЗЈЯрБШЃЌдкЗЧШмМСБЉТЖВаЛљЩЯБъМЧЕФПЩФмадДѓДѓНЕЕЭЃЌетИіР§згЭЙЯдСЫЕчЛЏбЇЙІФмЛЏЗНЗЈгыЯжгае§НЛЩњЮяХМСЊММЪѕЕФМцШнадЁЃ
ЮоЖРгаХМЃЌРзАЎЮФПЮЬтзщдк 2019 ФъБЈИцСЫвЛжжИХФюРрЫЦЕФРвАБЫсаоЪЮЗНЗЈ[27]ЃЌМДРћгУЕчЛЏбЇЗНЗЈЛюЛЏЗдрчрКЃЈPTZЃЉбмЩњЮяЃЈЭМ 3ЃЉЁЃетжжЗНЗЈПЩПьЫйЛёЕУвЛЯЕСагаМлжЕЕФЙІФмаоЪЮыФКЭЕААзжЪЃЌАќРЈв§ШыКЌЩњЮяЫиКЭФђЫсвЉЮяБћЛЧЪцЕФЗдрчрКЁЃШЛЖјЃЌИУЗНЗЈЖдКЌАыызАБЫсЕФЖўыФВЛФЭЪмЃЌЧвзЊЛЏЙ§ГЬжаЪЙгУСЫввыцКЭЫЎЛКГхвКЃЈ1:1ЃЉЕФЛьКЯЮяЃЌПЩФмЛсЯожЦИУЗНЗЈЕФЩњЮяЯрШнадвдМАдкИїжжЕААзжЪАаБъЩЯЕФЙуЗКгІгУЁЃЭтЃЌОЁЙмЛЏбЇбЁдёадКмИпЃЌЕЋзюГѕЕФe-Y-click КЭ PTZ ЗНЗЈЖМШБЗІЧјгђбЁдёадЃЌдкКЌгавЛИівдЩЯРвАБЫсВаЛљЕФЕзЮяжаЙлВьЕНЖржжаоЪЮЁЃвђДЫЃЌетаЉЗНЗЈЮоЗЈПЫЗўРЇШХЯжгаЩњЮяХМСЊММЪѕЕФЬивьадЮЪЬтЃЌВЂЭЙЯдСЫашвЊЕМЯђЛљРДИЈжњЮЛЕубЁдёадЙІФмЛЏЕФШБЕуЁЃ

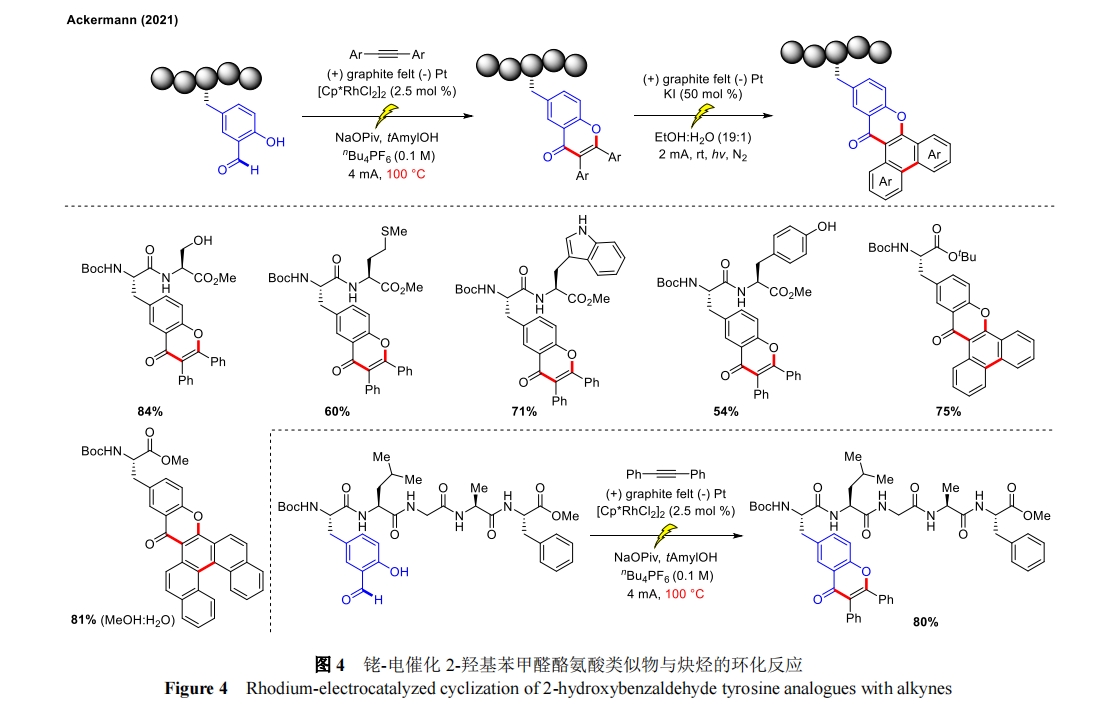
2021 ФъЃЌAckermann ПЮЬтзщБЈЕРСЫЕчЛЏбЇгыЙ§ЖЩН№ЪєююДпЛЏМСЯрНсКЯЃЌЖд 2-єЧЛљБНМзШЉРвАБЫсРрЫЦЮяКЭШВЬўНјааЛЗЛЏЗДгІ[28]ЃЌЙтЕчЛЏбЇбѕЛЏНщЕМЕФНјвЛВНАБЛљЫсаоЪЮПЩвдЛёЕУ Іа-РЉеЙыФЃЈОпгаЧПСвЕФгЋЙтЃЉЃЌДгЖјЙЙНЈФтыФКЭгЋЙтыФЁЃГ§СЫжЦБИИїжжаоЪЮЕФЖўыФЭтЃЌЛЙгУЖўБНЛљввШВдкєЧЛљБНМзШЉРвАБЫсРрЫЦЮяЩЯЖдЮхыФЕзЮяНјааСЫЙйФмЛЏЃЌвд 80%ЕФЗжРыВњТЪЕУЕНСЫЯргІЕФЩЋЭЊЃЈЭМ 4ЃЉЁЃИУЗДгІЫфШЛгЩШЉЩЯЕФМзѕЃЛљ-CЈDH ЛюЛЏв§ЗЂЃЌЕЋЗДгІЛњРэжаЕФЙиМќЕчЛЏбЇВНжшАќРЈ Rh(III/IV/II)бѕЛЏгеЕМЕФЛЙдЙ§ГЬЁЃживЊЕФЪЧЃЌгыЛЏбЇбѕЛЏМСЯрБШЃЌетжжЁАюю-ЕчДпЛЏЁБММЪѕФмИќгааЇЕиЛёЕУШЁДњЩЋЭЊЃЌеЙЯжГігХвьЕФЧјгђбЁдёадКЭНЯЙуЕФАБЛљЫсЕзЮяШнШЬадЁЃ
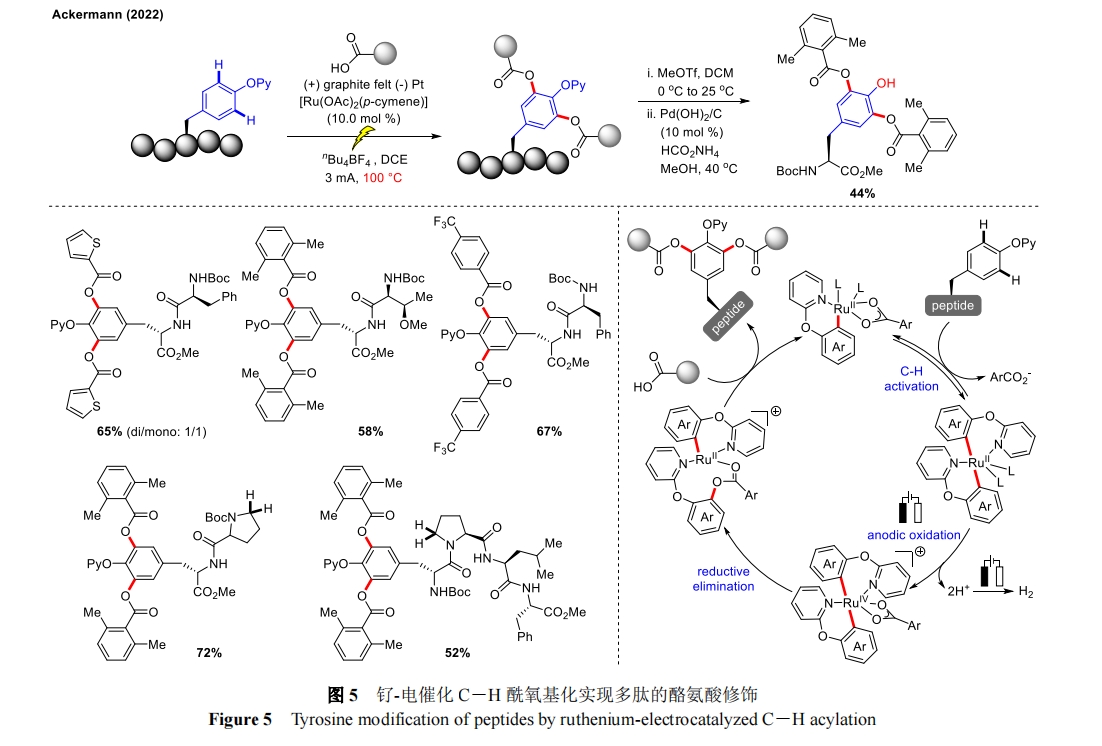
ЫцКѓЃЌAckermann ЭХЖггжЪЕЯжСЫюЩ-ЕчДпЛЏ CЈDH ѕЃбѕЛљЛЏгУгкРвАБЫсыФЕФКѓЦкаоЪЮ[29]ЃЌЗгєЧЛљБЃЛЄЛљКѓајПЩЭбГ§ЃЈЭМ 5ЃЉЁЃЕБвдЛЏбЇбѕЛЏМСШЁДњЕчзгНјааЗДгІЪБВЂЮДЛёЕУФПБъВњЮяЃЌЫЕУїЕчЛЏбЇЛЗОГБЃжЄСЫКѓЦкЙІФмЛЏЕФПЩГжајадКЭЮШЖЈадЁЃживЊЕФЪЧдкКЌгаИЌАБЫсЕФЖўыФЛђЫФыФЕзЮяжаЃЌИЌАБЫсЕФ ІС ЮЛВЂЮДЗЂЩњ Shono бѕЛЏЩњГЩЖўДЮаоЪЮЕФВњЮяЃЌЬхЯжГіСМКУЕФЮЛЕубЁдёадЁЃЛњРэбаОПжаЭЈЙ§ЯъЯИЕФ CV баОПКЭ DFT МЦЫуЮЊЫЋЛЗН№ЪєЛЏТчКЯЮяЕФаЮГЩвдМА Ru(II/IV/II)бѕЛЏгеЕМЕФЛЙдЙ§ГЬЬсЙЉСЫЧПгаСІЕФжЇГХЁЃИУВпТдЭЈЙ§ЖдРвАБЫсбмЩњыФНјааФЃПщЛЏКѓЦкѕЃбѕЛљЛЏЃЌЬсЙЉСЫЛёШЁДѓСПаоЪЮыФЕФБуНнЭООЖЃЌЭЛГіСЫЕчЛЏбЇКЭЙ§ЖЩН№ЪєДпЛЏжЎМфЕФаЭЌДпЛЏзїгУЁЃ
ШЛЖјдкИпЮТЃЈ100 ЁуCЃЉЯТНјааЕчНтПЩФмЛсЯожЦетРрЗНЗЈгУгкОпгавзБфадЕФЖўМЖЛђШ§МЖНсЙЙЕФДѓаЭЕААзжЪЕзЮяЃЛгыДѓЖрЪ§Н№ЪєДпЛЏаоЪЮВпТдвЛбљЃЌююЛђюЩЕФЪЙгУвВПЩФмЪЙКѓајДПЛЏИДдгЛЏЃЌЛђгЩгкЗЧЬивьадНсКЯЯрЛЅзїгУЖјЕМжТыФЪЇЛюЁЃ
зюНќЃЌИпУЮПЮЬтзщдкЧАШЫЙЄзїЕФЛљДЁЩЯБЈЕРСЫвЛжжЭЈЙ§жБНгЕчЛЏбЇЛюЛЏЗгєЧЛљРДДйНјКЌРвАБЫсЩњЮяДѓЗжзггыСкАБЛљБНМзЫсбмЩњЮязЊЛЏЕФЗНЗЈ[30]ЃЈЭМ 6AЃЉЁЃИУЗНЗЈРћгУЗњвѕРызгзїЮЊЕчНтжЪКЭЧтМќЬэМгМСЃЌгыРвАБЫсЗгєЧЛљжЎМфаЮГЩЧтМќЃЌФмЙЛКмКУЕиБъМЧРвАБЫсВаЛљЃЌОпгаКмИпЕФЛЏбЇКЭЮЛЕубЁдёадвдМАГіЩЋЕФзЊЛЏТЪЁЃдкЮТКЭЕФЛКГхЬѕМўЯТЃЌИїжжыФЁЂвЉЮяКЭЬьШЛВњЮяЖМОпгаКмКУЕФМцШнадЁЃДЫЭтЃЌМЁКьЕААзвВФмБЛЩњЮяШОЩЋМСЕхКьЫГРћБъМЧЧвВЛЛсЗЂЩњЗжНтЁЃЭЈЙ§ПижЦЪЕбщЁЂHR-MSЁЂ1H NMR КЭбЛЗЗќАВЗЈЃЈCVЃЉдкФкЕФЛњРэбаОПКЯРэЕиНтЪЭСЫРвАБЫсВаЛљжБНгбЁдёадЕчЛЏбЇЛюЛЏЕФЛњжЦЃЈЭМ 6BЃЉЁЃЪзЯШЃЌдкЗњвѕРызгЕФТчКЯзїгУЯТЃЌРвАБЫсдквѕМЋБЛЛЙдЪЭЗХ H2ЃЌЩњГЩБНЗгИКРызгжаМфЬхЁЃЫцКѓЕхКьгыБНЗгИКРызгжаМфЬхЗЂЩњЧзКЫМгГЩЗДгІЃЌЩњГЩбѕИКРызгжаМфЬхЁЃдк H2O ЕФЧзКЫзїгУЯТЗЂЩњПЊЛЗЗДгІЩњГЩДјгаєШЛљЕФжаМфЬхЃЌЫцКѓдкбєМЋбѕЛЏЭбєШЩњГЩзюжеВњЮяЁЃ
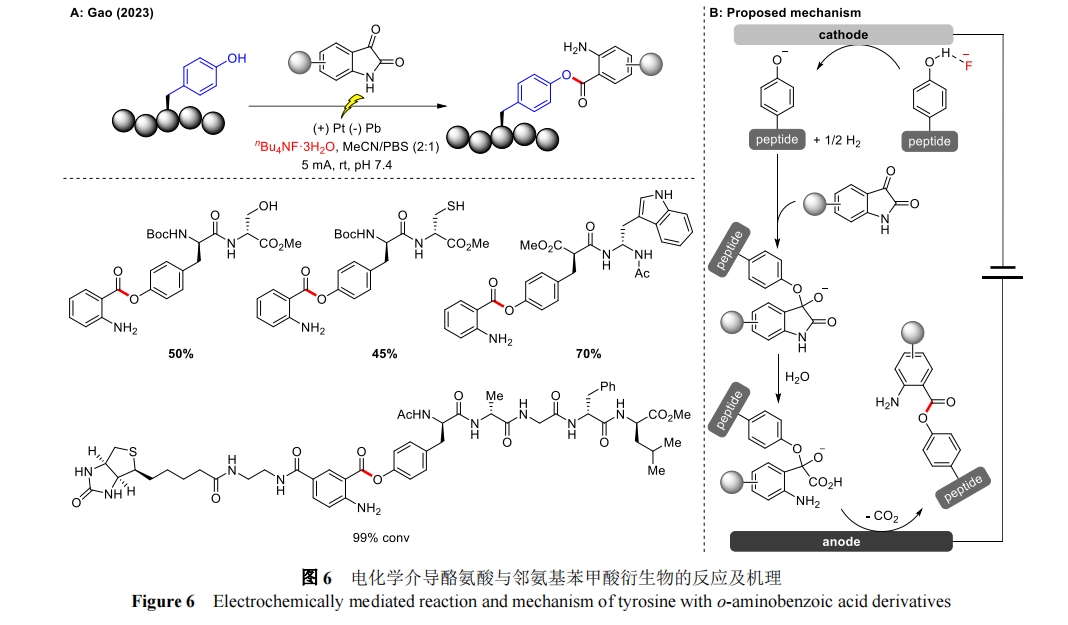
1.1.2 ЩЋАБЫс
дкЧАШЫЕФЛљДЁЩЯЃЌ2019 Фъ Kanai ПЮЬтзщЬсГіСЫвЛжжгУгкЖрыФКЭЕААзжЪЕФМфНгаоЪЮЗНЗЈ[31]ЃЌУшЪіСЫРћгУдЮЛЩњГЩЕФбѕАБЪдМСЖдЩЋАБЫсЕФЕчЛЏбЇаоЪЮЁЃетЯюЙЄзїЕФЙиМќЧјБ№дкгкаЁЗжзгЪдМСЕФЕчЛЏбЇЛюЛЏБЛШЯЮЊЪЧдкЪдМСгыЕААзжЪНсКЯКѓЗЂЩњЕФЃЌетгыЩЯЪіРвАБЫсЕФаоЪЮЗНЗЈВЛЭЌЃЈЭМ 7ЃЉЁЃИУЗНЗЈРћгУЯжгаЕФЩњЮяХМСЊВпТд[32]ЃЌЮоашЛЏбЇбѕЛЏМСКЭЬэМгМСЃЌЬѕМўЮТКЭЃЌБмУтСЫЭтВПбѕЛЏМСВЮгыЕФИБЗДгІЁЃПЩФмЕФЛњРэНвЪОГіЩЋАБЫсВрСДдкЩњЮяХМСЊЗДгІЕФЕчЛЏбЇВНжшжаЦ№ЕНСЫЙиМќзїгУЃЌЦфЕїНкСЫОпгаЕчЛюадЕФЕЊбѕЛЏЮяздгЩЛљЃЈЭЊ-ABNOЃЉЕФбѕЛЏЕчЮЛЃЌЪЙЭЊ-ABNO КЭЩЋАБЫсВрСДжЎМфаЮГЩвЛжжЁАдЄЙВщюИДКЯЮяЁБЃЌзюжеетжжИДКЯЮяЕФбѕЛЏЕчЮЛУїЯдЕЭгкЭЊ-ABNO ЛђЩЋАБЫсБОЩэЁЃвђДЫЃЌИУИДКЯЮяПЩжБНгдкбєМЋбѕЛЏЛђдкЕчЛЏбЇНщжЪЃЈШч 4-O-TEMPOЃЉжЎКѓБЛбѕЛЏЁЃВЛНіШчДЫЃЌЛюЛЏЕФИДКЯЮяПЩвдМЬајЗЂЩњЗДгІЃЌаЮГЩЭЊABNO ЙйФмЛЏЩњЮяХМСЊЮяЁЃжЕЕУзЂвтЕФЪЧЃЌИУЗДгІФмЙЛдкЫЎШмвКжаНјааЃЌПЩвдгыОпгабѕЛЏЛЙдЛюадЕФАБЛљЫсВаЛљЃЈгЮРыАыызАБЫсГ§ЭтЃЉМцШнЃЌВЂгІгУгкШмОњУИКЭХЃбЊЧхАзЕААзЬхЯЕЁЃ
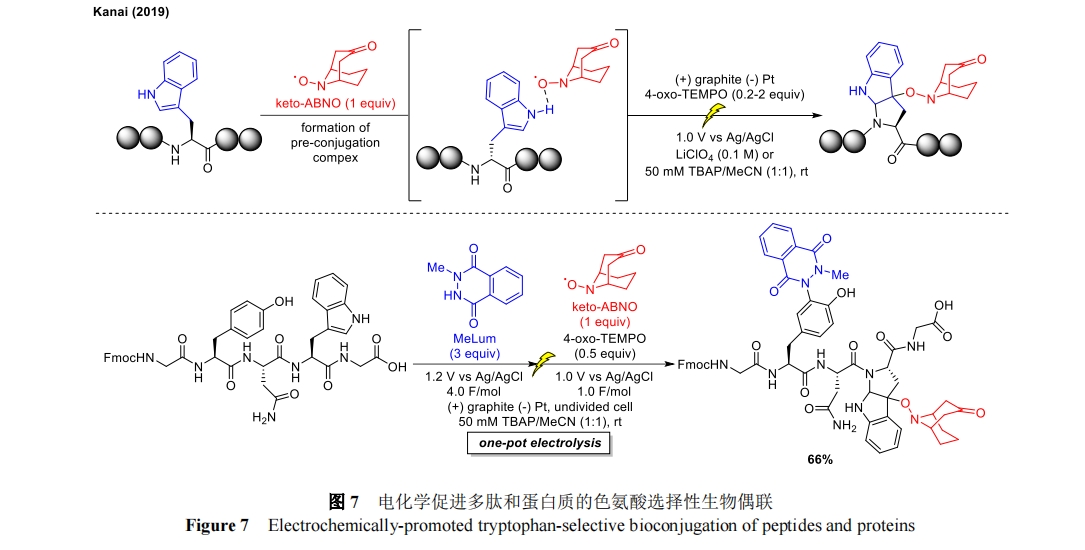
ДЫЭтЃЌетжжЗНЗЈЛЙеЙЪОСЫШчКЮРћгУЕчЛЏбЇдРэЃЌЭЈЙ§ЕїећЭтМгЕчбЙКЭЗДгІЬѕМўЃЌЧсЫЩЭъГЩСЌајЕФе§НЛЩњЮяХМЗДгІЁЃШчЭМ 7 ЫљЪОЃЌБЛ Fmoc БЃЛЄЕФЮхыФЭЈЙ§ e-Y-click ЗДгІЃЈМћЭМ 2ЃЉдкРвАБЫсЃЈдк 1.2 V ЯТЪЙгУЕчЛЏбЇЛюЛЏЕФ N-МзЛљТГУзХЕЃЉКЭЩЋАБЫсЃЈдк 1.0 V ЯТЪЙгУЭЊ-ABNO/4-O-TEMPO ЗНЗЈЃЉЩЯж№ВННјааЙІФмЛЏЁЃетаЉР§згЭЙЯдСЫЕчЛЏбЇММЪѕдкЛёШЁОпгаЖржиаоЪЮЕФыФКЭЕААзжЪЗНУцЕФЧБСІЁЃ
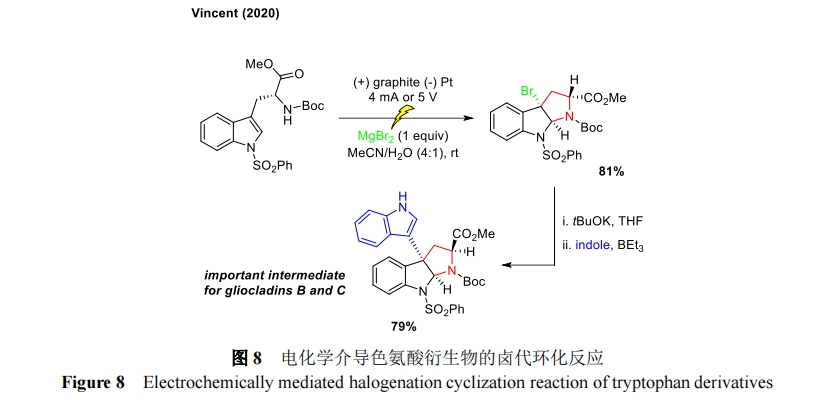
СэЭтЃЌVincent аЁзщдк 2020 ФъБЈЕРСЫвЛжжИпаЇЛЗБЃЕФЕчЛЏбЇЗНЗЈгУгкЩЋАБЫсбмЩњЮяЕФЭбЗМЛљТБДњЛЗЛЏ[33]ЃЌЫљЛёЕУЕФ 3a-фхпСПЉпХпспјЖдЬьШЛВњЮяШЋКЯГЩОпгаживЊвтвхЁЃетжжШЅЗМЙЙЛЏЙ§ГЬвРРЕгкЭЈЙ§ MgBr2 ЕФЕчЛЏбЇбѕЛЏВњЩњЧзЕчТБЫижаМфЬхЃЌЭЌЪБ MgBr2 дкИУЗДгІжазїЮЊЕчНтжЪБмУтСЫЦфЫћЕчНтжЪЕФЪЙгУЁЃвдВЛЭЌБЃЛЄЕФ L КЭ D-ЩЋАБЫсбмЩњЮяНјаафхЛЗЛЏЗДгІЃЌЗжБ№ЕУЕНСЫЯргІЕФВњЮяЁЃЭЈЙ§ X ЩфЯпОЇЬхНсЙЙжЄУїСЫЛЗЛЏЙ§ГЬОпгаЭтЯдбЁдёадЁЃДг D-ЩЋАБЫсЛёЕУЕФЭт-3a-фхпСПЉпХпспјПЩвдЭЈЙ§дк t-BuOK КЭШ§ввЛљХ№ЭщЕФДцдкЯТв§ШыпХпсНЋЦфзЊЛЏЮЊФк-3a-пХпс-пСПЉпХпспјЃЌетЪЧЩёОНКжЪПЫТЁЕААз B КЭ C КЯГЩЕФживЊжаМфЬхЃЈЭМ 8ЃЉЁЃ
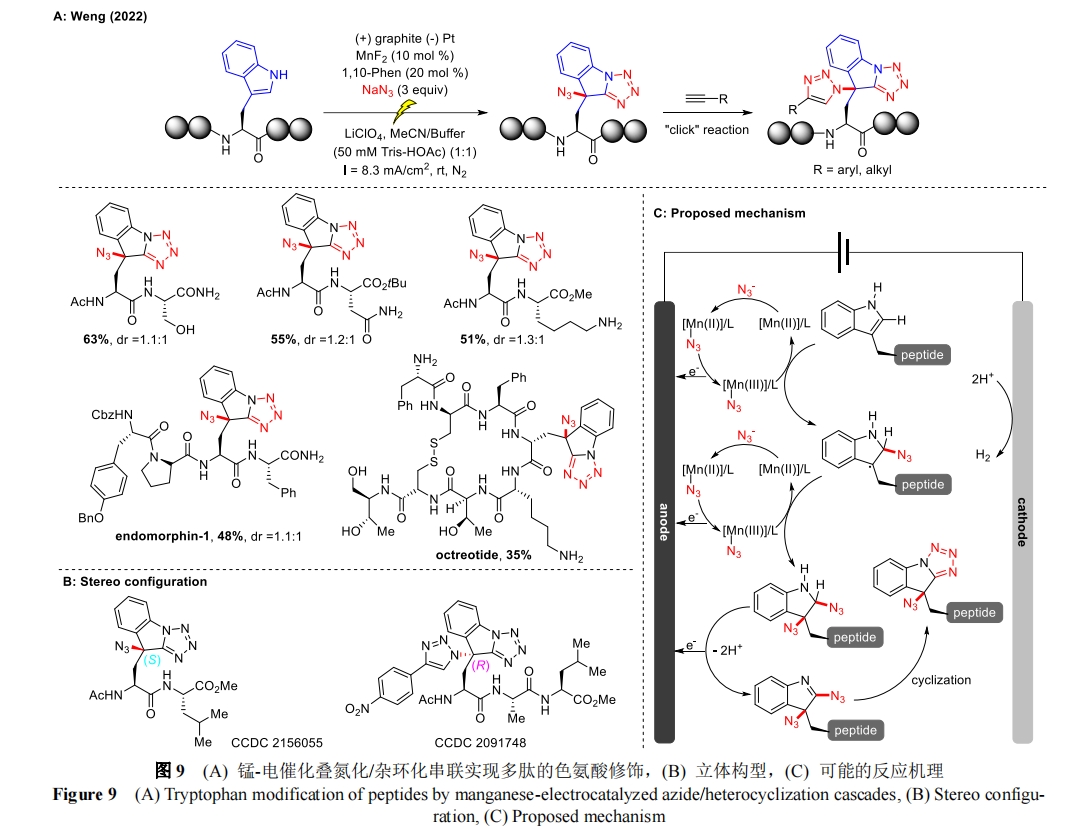
2022 ФъЃЌЮЬвтвтПЮЬтзщБЈЕРСЫвЛжжЭЈЙ§УЬДпЛЏДЎСЊздгЩЛљЕўЕЊЛЏ/дгЛЗЛЏЖдКЌгаЩЋАБЫсВрСДЕФыФНјааКѓЦкЙІФмЛЏЕФЕчЛЏбЇЗНЗЈ[34]ЃЈЭМ 9AЃЉЁЃетжжЕчЛЏбЇбѕЛЏВпТдЬсЙЉСЫЕўЕЊЛљШЁДњЕФЫФпђВЂ[1,5-a]пХпсЕФЖрыФбмЩњЮяЃЌОпгаЙуЗКЕФЙйФмЭХФЭЪмадКЭИпЮЛЕубЁдёадЃЌдкЮТКЭЕФЛКГхЬѕМўЯТвРШЛФмЕУЕНСМКУЕФЪеТЪЃЈИпДя 87%ЃЉЁЃВЛНіШчДЫЃЌЖдКЌгаЖржжУєИаАБЛљЫсЕФЖрыФЕзЮяЃЌШчЫПАБЫсЁЂРЕАБЫсЁЂЫеАБЫсвдМАЬьЖЌАБЫсЕШЃЌИУЕчЛЏбЇЬѕМўЯТШдШЛЪЧЯрШнЕФЁЃЭЈЙ§ИУЗНЗЈАЂЦЌРрыФФкЗШыФ-1ЁЂвЉЮяАТЧњыФвВФмГЩЙІЕиНјааКѓЦкНсЙЙаоЪЮЁЃИќживЊЕФЪЧЃЌдк C3 ЮЛБЛЕўЕЊЛљЭХаоЪЮЕФЩЋАБЫсыФПЩвдЭЈЙ§ЁАЕуЛїЁБЗДгІНјвЛВНбмЩњГівЛЯЕСаШ§пђВњЮяЃЌЮЊЙЙНЈИїжжбмЩњыФЦЬЦНСЫЕРТЗЁЃЭЈЙ§ X ЩфЯпОЇЬхбЇЗжЮіжЄУїСЫЕчЛЏбЇВњЮягыЖўДЮЁАЕуЛїЁБЗДгІзюжеВњЮяЕФЗЧЖдгГвьЙЙЬхЙЙаЭЃЈЭМ 9BЃЉЁЃЛњРэбаОПБэУїЩЋАБЫсыФЕФжиЕЊЛЏЪЧЭЈЙ§здгЩЛљМгГЩЭООЖЖјВЛЪЧздгЩЛљбєРызгЭООЖНјааЕФЃЌЭЈЙ§бЛЗЗќАВЗЈЃЈCVЃЉКЭздгЩЛљвжжЦЪЕбщЃЌКЯРэЕиНтЪЭСЫЕўЕЊздгЩЛљЕФЕчЛЏбЇЩњГЩЁЂздгЩЛљМгГЩвдМАЫцКѓЕФЭбЧтКЭдгЛЗЛЏЭООЖЃЈЭМ 9CЃЉЁЃ
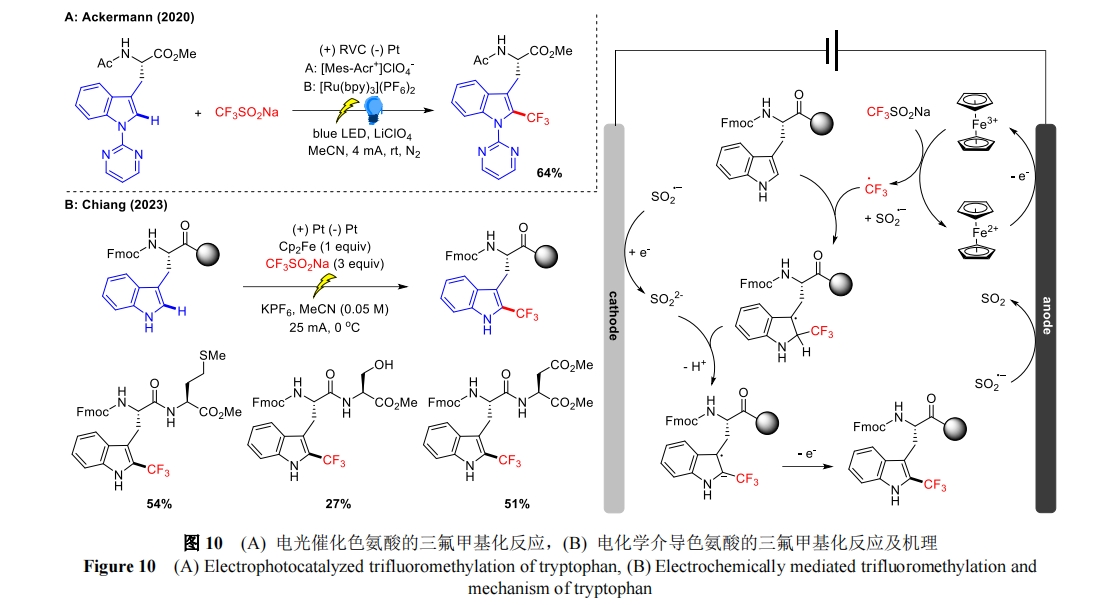
дкДЫжЎЧАЃЌ2020 ФъЃЌAckermann ПЮЬтзщОЭЭЈЙ§ЕчЙтаЭЌДпЛЏвд 64%ЕФВњТЪЪЕЯжСЫЩЋАБЫспХпсЛЗЩЯ C2 ЮЛЕФШ§ЗњМзЛљЛЏЃЈЭМ 10AЃЉ[35]ЃЌЕчКЯГЩгыЙтбѕЛЏЛЙдДпЛЏЕФНсКЯЮЊШ§ЗњМзЛљздгЩЛљЕФЩњГЩЬсЙЉСЫвЛжжЮоашЛЏбЇбѕЛЏМСЕФЭООЖЁЃНќЦкЃЌННЈОаЁзщдкИУЗНЗЈЕФЛљДЁЩЯБЈЕРСЫвЛжжМфНгЕчЛЏбЇВпТд[36]ЃЈЭМ 10BЃЉЃЌИУЗДгІвдЖўУЏЬњзїЮЊНщжЪЃЌЭЈЙ§ЕЅЕчзгзЊвЦЃЈSETЃЉЭООЖбѕЛЏ CF3SO2Na ВњЩњШ§ЗњМзЛљздгЩЛљЃЌЫцКѓШ§ЗњМзЛљздгЩЛљМгГЩЕНЩЋАБЫсВаЛљЩЯОЭбжЪзгЛЏЪЭЗХГіЯргІВњЮяЁЃИУЗНЗЈНіашЕчЛЏбЇДпЛЏЃЌЕзЮяЗЖЮЇгЩЕЅвЛЕФЩЋАБЫсЭиеЙЕНСЫЖўыФЕзЮяЃЌВЂЧвЩЋАБЫсЕФпХпсЛЗЮоашБЛБЃЛЄДгЖјЬсИпСЫВНжшОМУадЁЃЕЋвЛаЉКЌгаУєИаЛљЭХЕФЖўыФвзЗжНтЛђЪЇЛюЃЌНігаЩйСПЖўыФЕзЮяПЩвдЫГРћзЊЛЏЃЌЩњЮяМцШнадВюЃЌЯожЦСЫДЫЗНЗЈЕФгІгУЁЃ
1.1.3 БНБћАБЫс
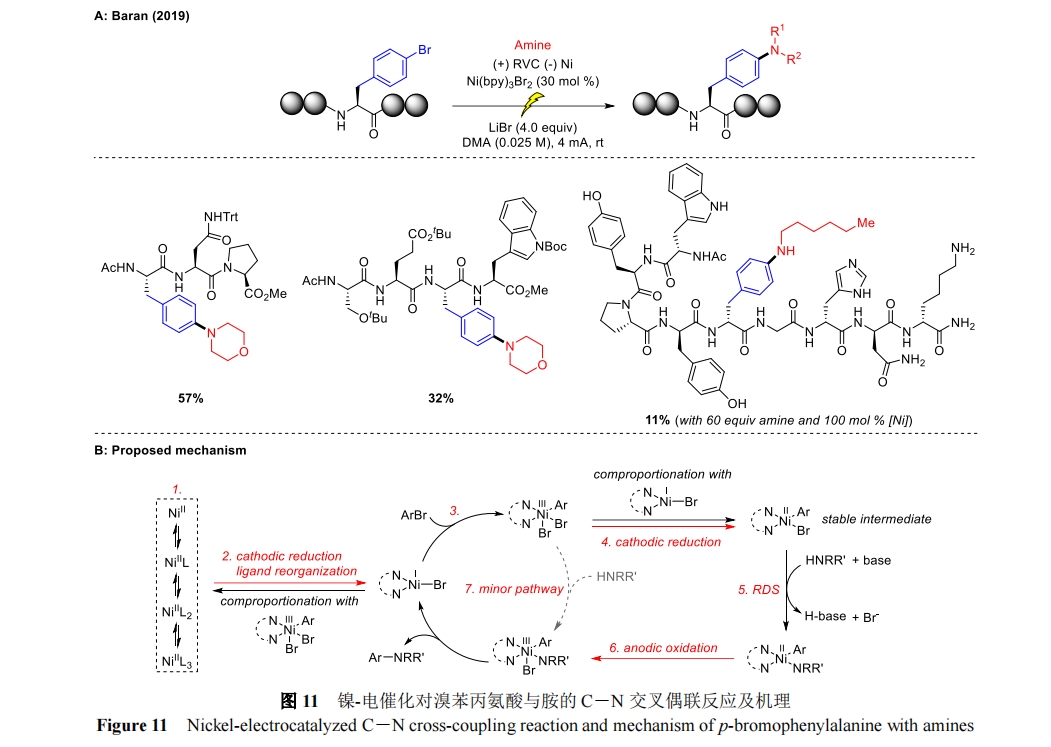
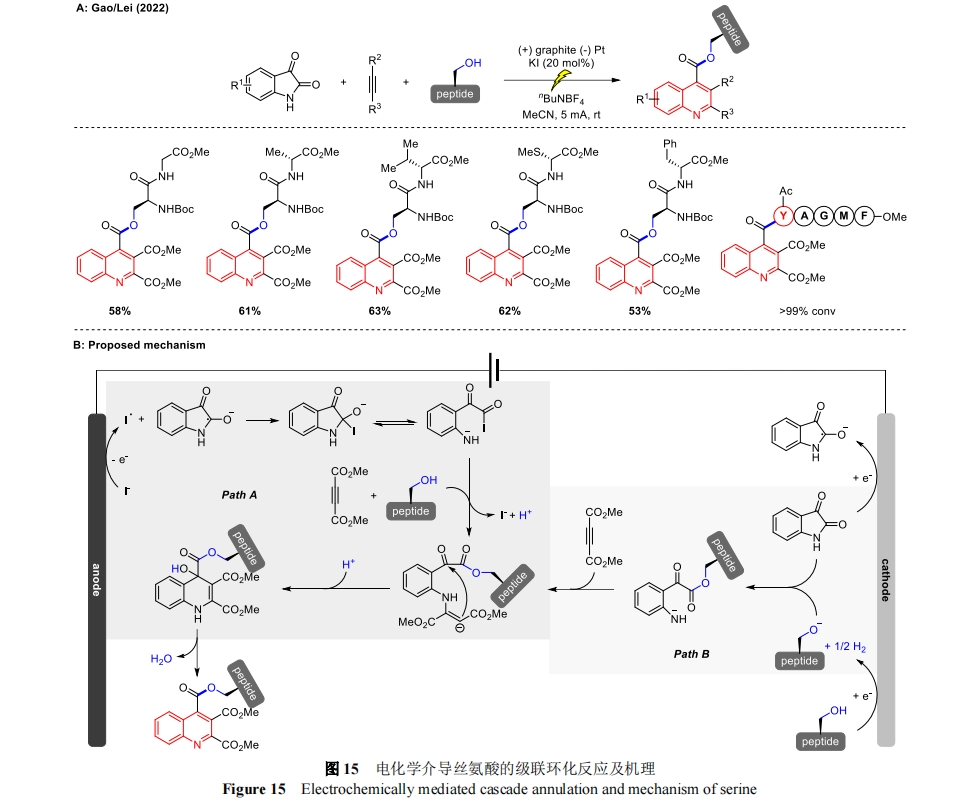
2019 ФъЃЌBaran ПЮЬтзщНЋФјДпЛЏгыЕчЛЏбЇЯрНсКЯЃЌЪЕЯжСЫЖдфхБНБћАБЫсМАШ§ыФЁЂЫФыФКЭЮхыФЕШгыАЗЕФ CЈDN НЛВцХМСЊЗДгІ[37]ЁЃДЫЭтЃЌЛЙЖдвЛжжЮДЪмБЃЛЄЕФШЩыФНјааСЫФјДпЛЏЕФЗМЛљТБАЗЛЏЃЌЮЊОпгабѕЛЏЛЙдЛюадЕФРвАБЫсКЭЩЋАБЫсВаЛљЬсЙЉСЫЫљашЕФАЗЛЏВњЮяЃЈЭМ 11AЃЉЁЃжЕЕУвЛЬсЕФЪЧЃЌетЪЧЕквЛИіЭЈЙ§ФјДпЛЏдкЙбыФЩЯаЮГЩ CЈDN МќЕФЪЕР§ЃЌОЁЙмЫќЕФВњТЪНЯЕЭЧвашвЊФјДпЛЏМСКЭЛЏбЇМЦСПЕФАЗХМСЊМСЁЃЛњРэбаОПжЄУїДпЛЏбЛЗЙ§ГЬжаЭЌЪБНјааСЫвѕМЋЛЙдКЭбєМЋбѕЛЏЁЃгЩ Ni(II)-дЄДпЛЏМСЕФвѕМЋЛЙдв§ЗЂЃЌаЮГЩ Ni(I)ЛюадДпЛЏМСЁЃШЛКѓЗМЛљфхЖд Ni(I)бѕЛЏМгГЩаЮГЩ Ni(III)ЃЌжЎКѓNi(III)БЛ Ni(I)вѕМЋЛЙдЩњГЩ Ni(II)жаМфЬхЁЃдкМюЕФзїгУЯТЃЌNi(II)жаМфЬхгыАЗЗЂЩњХфЬхНЛЛЛВЂдйДЮБЛбєМЋбѕЛЏЁЂЛЙдЯћГ§ЩњГЩЫљашЕФАЗЛЏВњЮяЃЈЭМ 11BЃЉЁЃЖдЩњЮягІгУЖјбдЃЌЗДгІжаЪЙгУЕФЖўМзЛљввѕЃАЗЃЈDMAЃЉКЭ LiBr ПЩФмашвЊЖюЭтЕФДПЛЏВНжшЃЌПЩФмЖдИУММЪѕЕФЙЄвЕЛЏгавЛЖЈгАЯьЁЃОЁЙмШчДЫЃЌИУЗНЗЈзїЮЊвЛжжзщзАИпМлжЕЖрыФ PTMsЃЈгШЦфЪЧгыДѓЛЗ RiPP ЯрЙиЕФЖРЬиНЛСЊЃЉЕФЗНЗЈЃЌЮЊЩњЮяДѓЗжзгЕФЬивьадаоЪЮЬсЙЉСЫПЩФмЁЃ2021 ФъвЖН№аЧПЮЬтзщдкЩЯЪіЗНЗЈЕФЛљДЁЩЯвд 73%ЕФВњТЪВЙГфСЫЖдфхБНБћАБЫсЕФАБМзЛљЛЏЪЕР§[38]ЃЌНјвЛВНЬНОПСЫИУЗДгІЕФЧБдкгІгУМлжЕЁЃ
1.1.4 ИЌАБЫс
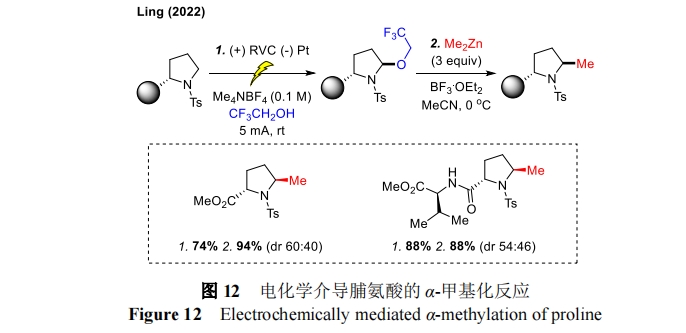
2022 ФъЃЌСжЫЩПЮЬтзщЛљгкОЕфЕФ Shono бѕЛЏБЈЕРСЫвЛжжИЌАБЫсЕФЮЛЕубЁдёад ІС-МзЛљЛЏЗНЗЈ[39]ЃЈЭМ 12ЃЉЁЃдкетЯюЙЄзїжаЃЌЪзЯШЭЈЙ§ЕчЛЏбЇЧ§ЖЏЕФ C-H ЛюЛЏЩњГЩ N,O-ЫѕШЉЛЏКЯЮяЃЌЫцКѓдкгаЛњаПЪдМСЕФНщЕМЯТНјвЛВНЩњГЩ ІС-МзЛљЛЏВњЮяЁЃгУШ§ЗњввДМЃЈTFEЃЉЬцДњГЃгУЕФввДМШмМСЛђЧзКЫЪдМСЃЌПЩвдЪЙЖшадЕзЮяИќШнвзБЛМЄЛюЁЃДЫЭтЃЌИУВпТдЛЙЪЕЯжСЫИїжжНсЙЙИДдгАЗбмЩњЮяЕФКѓЦкЙІФмЛЏЃЈLSFЃЉЃЌШчАБЛљМзЫсѕЅРрЁЂЛЧАЗРрКЭѕЃАЗРрЃЌОпгаЗДгІЗЖЮЇЙуЁЂЙйФмЭХМцШнадИпКЭВйзїМђЕЅЕШЬиЕуЁЃ
1.1.5 АыызАБЫс
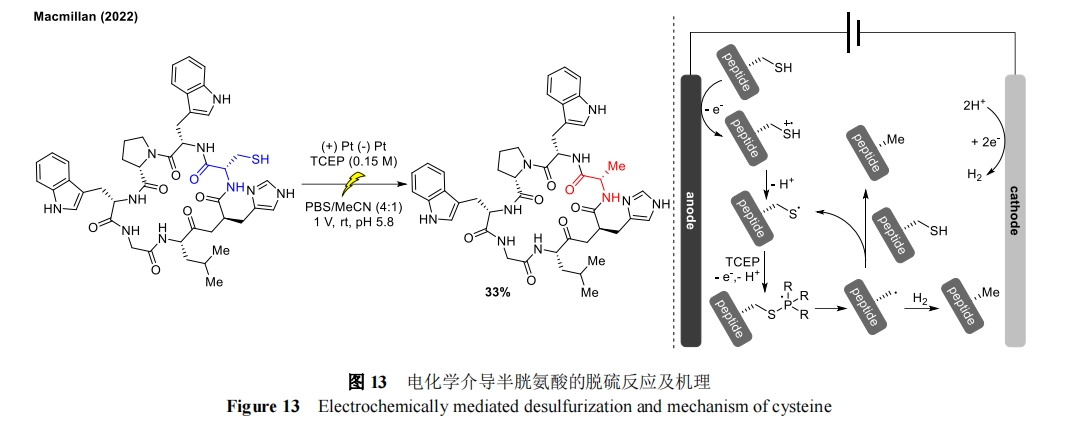
2022 ФъЃЌMacmillan аЁзщЭЈЙ§ЕчЛЏбЇбѕЛЏЪЕЯжСЫАыызАБЫсЕФЭбСђЗДгІ[40]ЁЃАыызАБЫсВаЛљОбєМЋбѕЛЏКѓЩњГЩздгЩЛљбєРызгЃЌШЅжЪзгЛЏаЮГЩЭщЛљСђздгЩЛљЁЃИУСђздгЩЛљдкШ§єШввЛљьЂЃЈTCEPЃЉЕФзїгУЯТаЮГЩьЂздгЩЛљжаМфЬхШЛКѓЗЂЩњ ІТ СбНтЃЌДгЖјВњЩњаТЕФЬМздгЩЛљЃЌЬМздгЩЛљПЩДгЦ№ЪМдСЯЛђвѕМЋВњЩњЕФЗжзгЧтжаО№ШЁЧтЪЕЯжЭбСђЃЌЭЌЪБЪЭЗХСђздгЩЛљЛиЕНДпЛЏбЛЗЯЕЭГжаЁЃЪЪгУЕФЕзЮяГ§АыызАБЫсЭтЃЌЛЙЪЕЯжСЫЖржжЖЬЛЗыФЕФЭбСђЗДгІЃЈЭМ 13ЃЉЁЃ
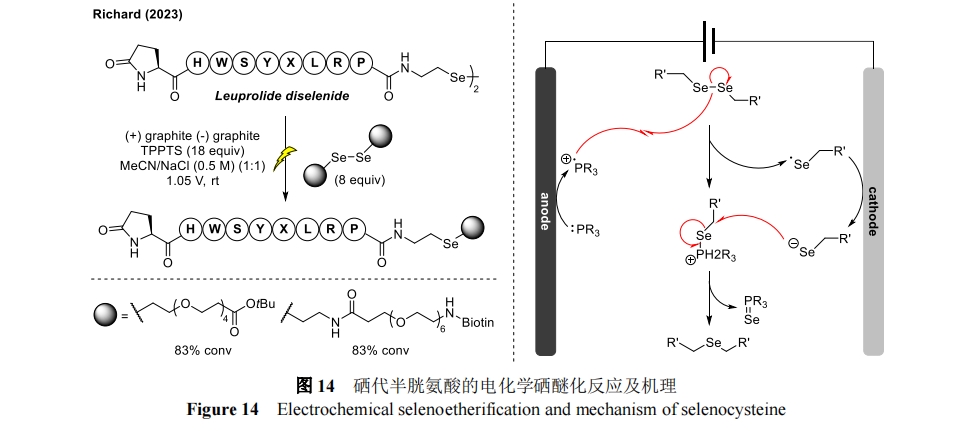
2023 ФъЃЌRichard ПЮЬтзщУшЪіСЫвЛжжЕчЛЏбЇЮјУбЛЏЃЈe-SEЃЉВпТдЃЌгУгкЖрыФЕФЮЛЕубЁдёадЙІФмЛЏ[41]ЁЃИУЗНЗЈЪмЙтДпЛЏЖўЮјЛЏЮяЪеЫѕЃЈPDCЃЉзЊЛЏЃЈЪЙгУвПЙтДпЛЏМСКЭьЂдк 45 nm ДІЗјееЃЌНЋВЛЖдГЦЛђЖдГЦЕФЖўЮјЛЏЮяЦ№ЪМВФСЯзЊЛЏЮЊЯргІЕФЮјУбСЌНгВњЮяЃЉЕФЦєЗЂЃЌРћгУЮјДњАыызАБЫсЕФЖРЬиЗДгІадЃЌдкЮТКЭЕФЕчЛЏбЇЬѕМўЯТЭЈЙ§ЮШЖЈЕФЮјУбМќаЮГЩгаМлжЕЕФЩњЮязККЯЮяЁЃЕчЛЏбЇПЩвдОЋзМЕФПижЦбѕЛЏЛЙдЕчЮЛЃЌПЫЗўСЫ PDC ЕФОжЯоадЃЌАќРЈгыгЋЙтЕзЮяВЛЯрШнвдМАЪЙгУАКЙѓКЭЫЎШмадВюЕФЙтДпЛЏМСЃЌЪЕЯжСЫЕЭГЩБОЁЂдзгОМУКЭЩњЮяЯрШнЕФЗНЗЈЖдЮјДњАыызАБЫсВаЛљНјааЮЛЕубЁдёадЙІФмЛЏЁЃE-SE ЕФФмСІЭЈЙ§ FDA ХњзМЕФПЙАЉвЉЮяССБћШ№СжЕФЭэЦк C ФЉЖЫаоЪЮЖјЭЛГіЯдЪОЁЃЛњРэбаОПжаЃЌьЂбєМЋбѕЛЏВњЩњЕФздгЩЛљбєРызгЛсгеЕМЖўЮјЛЏЮяОљСбЃЌгЩДЫВњЩњЕФЮјздгЩЛљОвѕМЋЛЙдЛсЩњГЩвЛЕБСПЕФИпЧзКЫадЮјЫсбЮЁЃШЛКѓЃЌетжжЛюадЮяжЪПЩвдЙЅЛїЖўЮјЛЏЮяСбНтЙ§ГЬжаВњЩњЕФЧзЕчЮјЛљьЂЃЌДгЖјЕУЕНЫљашЕФЮјУбСЌНгВњЮяКЭЮјЛЏСзИБВњЮяЃЈЭМ 14ЃЉЁЃ
1.1.6 ЫПАБЫс
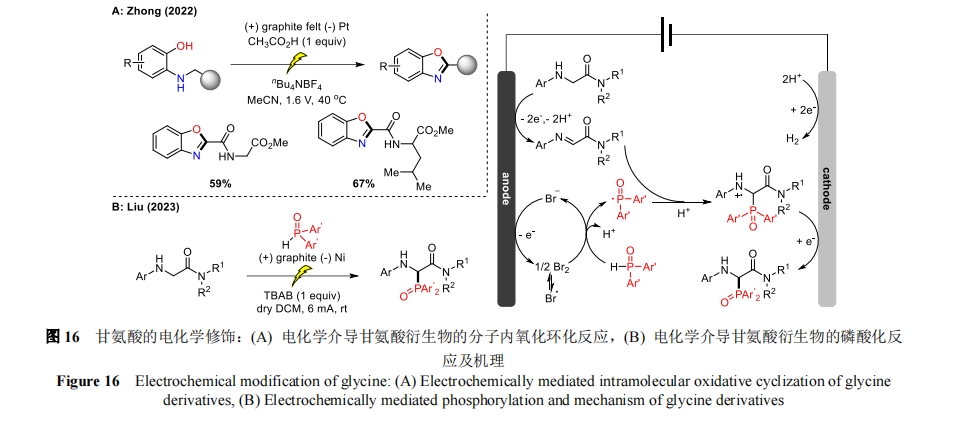
2022 ФъЃЌИпУЮКЭРзАЎЮФПЮЬтзщЙВЭЌБЈЕРСЫвЛжжХфЖдЕчНтжЇГжЕФМЖСЊЛЗЛЏЗНЗЈЃЌвдЕхКьЁЂШВЬўКЭЫПАБЫсЗжзгзїЮЊЦ№ЪМВФСЯИпаЇЕФКЯГЩСЫвЛЯЕСаИпЙІФмЛЏЕФрпјШЁДњЕФЩњЮяЛюадЗжзг[42]ЃЈЭМ 15AЃЉЁЃдкЮТКЭЕФ pH жаадЬѕМўЯТЃЌетжжзЊЛЏПЩвдФЭЪмЙуЗКЕФЙйФмЭХЃЌАќРЈЫПАБЫсВаЛљЁЂЖрыФЁЂЬЧРрЁЂвЉЮяКЭЬьШЛВњЮяЃЌвВПЩвдзїЮЊвЉЮябаОПКЭЦфЫћСьгђЕФЙІФмЛЏВпТдЁЃЭЈЙ§бЛЗЗќАВЗЈЃЈCVЃЉКЭПижЦЪЕбщЬсГіСЫПЩФмЕФЗДгІЛњРэЃЈЭМ 15BЃЉЁЃЪзЯШЃЌЕхКьдквѕМЋБЛЛЙдЩњГЩздгЩЛљвѕРызгЃЌЭЌЪБЃЌKI ОбєМЋбѕЛЏЩњГЩЕтздгЩЛљЃЌЕхКьздгЩЛљвѕРызггыЕтздгЩЛљНЛВцХМСЊЩњГЩбѕИКРызгжаМфЬхЁЃИУжаМфЬхОЙ§ПЊЛЗЗДгІКѓЃЌгыЫПАБЫсВаЛљКЭШВЬўЗжБ№ЗЂЩњЧзКЫШЁДњКЭМгГЩЗДгІЩњГЩЬМИКРызгжаМфЬхЃЌЕтвѕРызгдйЩњЭъГЩДпЛЏбЛЗЃЈТЗОЖ AЃЉЁЃДЫЭтЃЌЫПАБЫсВаЛљПЩФмдквѕМЋБЛЛЙдВњЩњЭщбѕРызгЃЌШЛКѓгыЕхКьЗДгІЩњГЩПЊЛЗЕЊИКРызгжаМфЬхЃЌетвЛЙ§ГЬжа KI зїЮЊбєМЋЮўЩќМСЃЈТЗОЖ BЃЉЁЃзюКѓОЗжзгФкЛЗЛЏЭбЫЎЩњГЩЯргІЕФрпјВњЮяЁЃ
1.1.7 ИЪАБЫс
2022 ФъЃЌжгЮЊЛлПЮЬтзщПЊЗЂСЫвЛжжЪЕгУЧвдзгОМУЕФЕчДпЛЏВпТд[43]ЃЌЭЈЙ§ Shono-аЭбѕЛЏХМСЊЖдИЪАБЫсбмЩњЮяНјааЗжзгФкбѕЛЏЛЗЛЏЃЈЭМ 16AЃЉЁЃгыжЎЧАЕФбаОПЯрБШЃЌИУЗНЗЈЮоашЬэМгбѕЛЏМСКЭН№ЪєДпЛЏМСЃЌдзгОМУадКУЃЌвджаЕШжССМКУЕФВњТЪКЯГЩЖржж 2-ШЁДњБНВЂЖёпђЃЌНіВњЩњ H2 зїЮЊИБВњЮяЁЃНќЦкЃЌСѕГПНПЮЬтзщЭЈЙ§ C(sp3)ЈDH ЙйФмЛЏЪЕЯжСЫИЪАБЫсбмЩњЮягыКЌ P(O)ЈDH/PЈDH ЕФЛЏКЯЮяЕФЕчЛЏбЇбѕЛЏСзЫсЛЏЗДгІ[44]ЃЈЭМ 16BЃЉЁЃИУЗНЗЈОпгаЙуЗКЕФЕзЮяЗЖЮЇКЭНЯИпЕФЙйФмЭХФЭЪмадЃЌНЋЗДгІЬѕМўЩдзїИФБфКѓПЩНјааПЫМЖЙцФЃЗДгІЁЃЛњРэбаОПжавд(E)-N,N-ЖўМзЛљ-2-ЃЈБНЛљбЧАБЛљЃЉввѕЃАЗДњЬцФЃАхЕзЮядкБъзМЬѕМўЯТНјааЗДгІЃЌПЩвдвд 56%ЕФВњТЪЕУЕНФПБъВњЮяЁЃдкБъзМЗДгІ 1h КѓЕФЗДгІвКжавВЭЈЙ§ HRMS МьВтЕНСЫФПБъВњЮяЃЌетаЉНсЙћБэУї(E)-N,N-ЖўМзЛљ-2-ЃЈБНЛљбЧАБЛљЃЉввѕЃАЗЪЧетвЛзЊЛЏЙ§ГЬжааЮГЩЕФживЊжаМфЬх.
1.2 ЦфЫћАБЛљЫсгыЖрыФВрСДЕФЕчЛЏбЇаоЪЮ
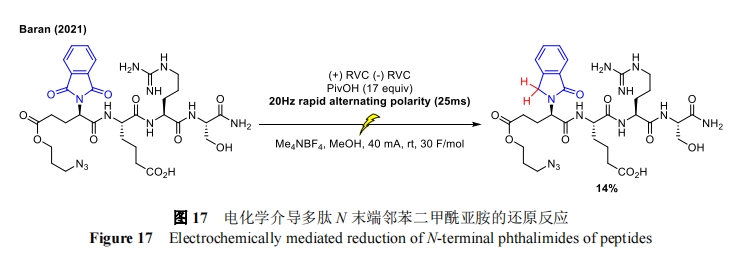
2021 ФъЃЌBaran ПЮЬтзщНвЪОСЫвЛжжНЛСїЕчНтаЮЪНЕФЪЕМЪгІгУ[45]ЃЌМДЪЙгУПьЫйНЛСїЕчЖјЗЧжБСїЕчЁЃПьЫйНЛЬцЕчМЋЃЈrAPЃЉПЩвдЧјЗждкЯрЭЌбѕЛЏЛЙдЕчЮЛЯТЗЂЩњЕФЛЏбЇЗДгІЫйТЪЃЌДгЖјДйНјЛЏбЇбЁдёадЕчКЯГЩЃЌЖјВЛНіНівРППЕчЛЏбЇЕчЪЦРДв§ЕМбѕЛЏЛЙдЛЏбЇЗДгІЁЃИУЗНЗЈЭЈЙ§бЁдёадЛЙд N ФЉЖЫСкБНЖўМзѕЃбЧАЗБЃЛЄЛљЭХЃЌЕУЕНЯргІЕФФкѕЃАЗЃЌГЩЙІЕФЪЕЯжСЫЫФыФФЃаЭЃЈКЌгаАЫИібѕЛЏЛЙдВЛЮШЖЈЮЛЕуЃЉЕФбЁдёадаоЪЮЃЈЭМ 17ЃЉЃЌЬхЯжГіЕчЛЏбЇЗДгІЕФИпЮЛЕубЁдёадЁЃжЕЕУзЂвтЕФЪЧЃЌЖрыФжаДцдкДѓСПЕФбѕЛЏЛЙдЛюадЙйФмЭХЃЌФмЙЛдкЯрЭЌЛђЯрЫЦЕчЪЦЯТбЁдёЬиЖЈЕФбѕЛЏЛЙдЗДгІЖјВЛЗЂЩњЦфЫћИБЗДгІЁЃЕЋЪЧЃЌгЩгкЪЙгУ MeOH зїЮЊШмМСЃЌВЂЧвВњТЪВЛИпЃЌвђДЫашвЊНјвЛВНПЊЗЂЃЌВХФмНЋетжжзЊЛЏЗНЗЈЭЦЙуЕНИќгаЪЕМЪвтвхЕФЖрыФЛђЕААзжЪЯЕЭГжаЁЃОЁЙмШчДЫЃЌетЯюбаОПвВГѕВНЬхЯжГіЕчЛЏбЇдкеце§бЁдёадаоЪЮыФКЭЕААзжЪЗНУцЕФЧБСІЁЃ
ДЫЭтЃЌШюжОалПЮЬтзщБЈЕРСЫвЛжжаТаЭЮјЕчДпЛЏ 2-ввЯЉЛљБНАЗЕФЭбЧтЛЗЛЏЗДгІ[46]ЃЌЭЈЙ§ CЈDH/NЈDH ЛюЛЏКЯГЩЙйФмЛЏпХпсЖрыФбмЩњЮяЁЃвдКЌгаИїжжАБЛљЫсЕФ 2-ввЯЉЛљБНАЗЮЊЕзЮяЃЌдкЖўЮјЛЏЮяДпЛЏЯТНјааЮТКЭЕФЕчбѕЛЏЛЗЛЏЗДгІЃЌГЩЙІЕиЛёЕУСЫвЛЯЕСапХпсбмЩњЮяЃЌЧвЮоЭтЯћа§ЛЏЯжЯѓЃЈЭМ 18ЃЉЁЃживЊЕФЪЧЃЌЫПАБЫсКЭРвАБЫсЕФгЮРыєЧЛљвдМАзщАБЫсЕФгЮРыАБЛљдкИУЕчЛЏбЇзЊЛЏжаБЛЫГРћБЃСєЃЌЖўыФЁЂШ§ыФКЭЫФыФЕзЮявВФмКмКУЕФБЛМцШнЁЃИУЗНЗЈЧПДѓЕФЭбЧтЛЗЛЏФмСІЃЌПЩЖдИДдгЕФЩњЮяЛюадЗжзгЬхЯЕНјааКѓЦкаоЪЮЃЌДгЖјЮЊЖрыФБъМЧаоЪЮпХпсЕФЖрЙІФмКЯГЩЕьЖЈСЫЛљДЁЁЃ
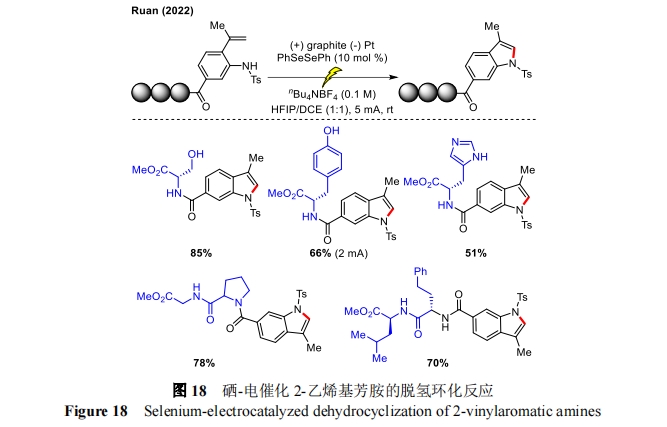
дчдк 2018 ФъЃЌГ№гбАЎПЮЬтзщОЭНЋвПДпЛЏгыЕчбѕЛЏЯрНсКЯЪЕЯжСЫКЌБНБћАБЫсЙЧМмЕФЯЉЬўгыБНМзЫсЕФЛЗЛЏ[47]ЃЈЭМ19AЃЉЁЃ2022 ФъЃЌЫћУЧгждйДЮБЈЕРСЫ N-ЗМЛљЛЗАЗЃЈКЌБНБћАБЫсЙЧМмЃЉЕФЕчЛЏбЇШЅБЅКЭ ІТ ѕЃЛЏЗДгІ[48]ЃЈЭМ 19BЃЉЁЃЫцКѓгжвдМђЕЅЕФ D2O зїЮЊыЎдДЃЌЪЕЯжСЫЭщЛљТБЛЏЮяЃЈКЌБНБћАБЫсКЭчгАБЫсЙЧМмЃЉЕФЕчЛЙдыЎЛЏЗДгІ[49]ЃЌыЎЩјШыТЪИпДя99%ЃЈЭМ 19CЃЉЁЃЭЌФъЃЌИУПЮЬтзщЛЙБЈЕРСЫвЛжжМђБуИпаЇЕФЗНЗЈжЦБИОпгаживЊКЯГЩМлжЕЕФ ІТ-єЧЛљЫсбмЩњЮя[50]ЃЈЭМ 19DЃЉЁЃИУЗДгІРћгУЗМЛљЛЗбѕЛЏЮяКЭЖўбѕЛЏЬМЮЊЕзЮяЃЌдкЮТКЭЁЂПЩГжајЕФЕчЛЏбЇЬѕМўЯТЪЕЯжСЫ ІТ-єЧЛљЫсЕФбЁдёадКЯГЩЁЃИУЗНЗЈЕзЮяЗЖЮЇЙуЃЌЙйФмЭХМцШнадКУЃЌвЛЯЕСаКЌгаАБЛљЫсЙЧМмЕФЗМЛљЛЗбѕЛЏЮяОљФмзЊЛЏЮЊгагУЕФєЧЛљЫсЁЃДЫЭтЃЌЖдИДдгЗжзгКЭвЉЮябмЩњЮяЕФНјвЛВНКѓЦкаоЪЮжЄЪЕСЫИУЗНЗЈЕФЧБдкгІгУЁЃЛњРэбаОПИљОнЗМЛљЛЗбѕЛЏЮяЕФЛЙдФмСІЬсГіСЫСНжжПЩааЭООЖЁЃЭООЖ A ЗМЛљЛЗбѕЛЏЮяжБНгЗЂЩњЕчЛЏбЇЛЙдЬсЙЉздгЩЛљИКРызгжаМфЬхЁЃЭООЖ B дђЪЧЭЈЙ§ CO2 ЕФЕЅЕчзгЛЙдЙ§ГЬЕУЕНвЛИіЕчзгКѓдйЩњГЩздгЩЛљИКРызгжаМфЬхЁЃИУжаМфЬхНјаа CЈDO МќСбНтЃЌЕУЕН C-жааФздгЩЛљЁЃЫцКѓОЙ§СэвЛДЮЕЅЕчзгЛЙдКЭ CO2 ЕФ C-ЧзКЫНјЙЅЃЌзюКѓдкбЮЫсЫЎШмвКЕФзїгУЯТЩњГЩ ІТ-єЧЛљЫсЁЃ
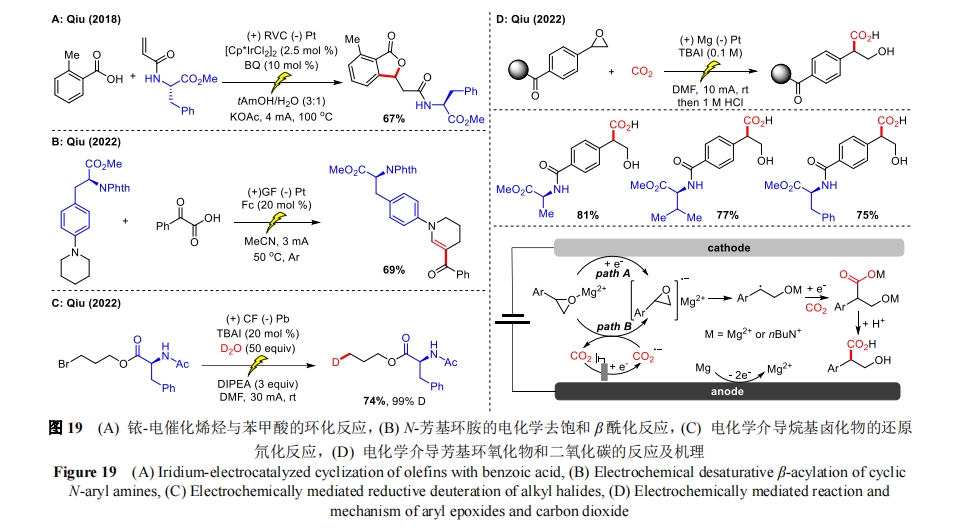
2023 ФъЃЌаЛОЂПЮЬтзщБЈЕРСЫдкЕчЛЏбЇЬѕМўЯТЃЌгЩЫЋКЫН№ДпЛЏНсЙЙЖрбљЕФШВЬўгыЗМЛљыТжЎМфЕФ C(sp)-C(sp2)ХМСЊЗДгІ[51]ЃЌЬсЙЉСЫвЛжжЪЕгУЕФ C-C бѕЛЏХМСЊЗНЗЈЃЈЭМ 20AЃЉЁЃИУЗНЗЈЖдКЌгаУєИаЙйФмЭХЕФДМРрЁЂАЗРрЁЂСђЛЏЮяКЭИЛЕчзгЯЉРрЕШОпгаМЋМбЕФМцШнадЃЌВЂЧвгыАБЛљЫсЁЂЖрыФЁЂКЫмеЫсКЭЬЧЕШЩњЮяДѓЗжзгЯрСЌЕФШВЬўвВФмГЩЙІБЛаоЪЮЃЌЭЛГіСЫИУЗНЗЈЕФКЯГЩЮШЖЈадЁЃЛњРэбаОПБэУїЃЌЗМЛљыТЪзЯШдкбєМЋБЛбѕЛЏаЮГЩИпЧзЕчадЗМЛљздгЩЛљЃЌИУздгЩЛљПЩвдИњ Au(I)ДпЛЏМСЩњГЩ Au(II)ЮяжжЃЌШЛКѓОбєМЋбѕЛЏВњЩњИпЧзЕчадЕФ Ar-Au(III)жаМфЬхЃЌШЛКѓгыФЉЖЫШВЬўаЮГЩШВЛљ Au(III)ЃЌзюКѓОЛЙдЯћГ§ЪЭЗХГіХМСЊВњЮяЃЈЭМ 20BЃЉЁЃ
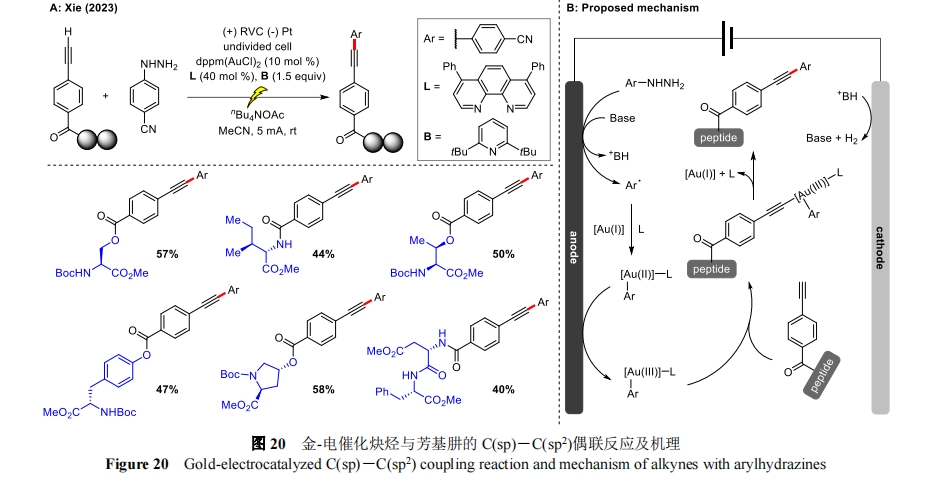
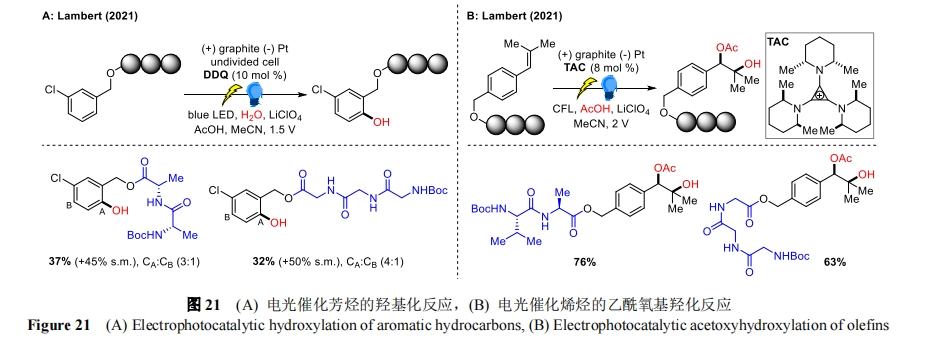
2021 ФъЃЌLambert ПЮЬтзщЪЙгУ 2,3-ЖўТШ-5,6-ЖўЧшЛљѕЋЃЈDDQЃЉзїЮЊЕчЙтДпЛЏМСЃЌПЊЗЂСЫвЛжжОпгаИпЛЏбЇбЁдёадЕФЗМЬўєЧЛљЛЏЗНЗЈ[52]ЃЌПЩвдНЋЫЎЁЂДМЁЂЫсЁЂѕЃАЗЛђАБЛљМзЫсѕЅЬэМгЕНЗМЬўРрЛЏКЯЮяжаЃЌЮоашЪЙгУЭтВПбѕЛЏМСЃЌгыЖрыФЯрСЌЕФЗМЬўвВФмЫГРћзЊЛЏЃЈЭМ 21AЃЉЁЃЛњРэбаОПБэУїЃЌИУЙ§ГЬЭЈЙ§ DDQ ЕФдйбЛЗРДЪЕЯжЃЌЦфжаЙтМЄЗЂЕФ DDQ ОЕЅЕчзгзЊвЦЃЈSETЃЉбѕЛЏЗМЬўЃЌДгЖјЬсЙЉвЛИіПЩБЛЧзКЫЪдМСВЖЛёЕФздгЩЛљбєРызгЁЃЭЌФъЃЌЫћУЧЛЙБЈЕРСЫвЛжжЕчЙтДпЛЏВпТдЃЌВЩгУШ§АБЛљЛЗБћЯЉЃЈTACЃЉРызгДпЛЏМСЃЌдкПЩПиЕФЕчЛЏбЇЕчЮЛЯТЃЌЪЕЯжСЫЯЉЬўЕФввѕЃбѕЛљєЧЛЏЗДгІ[53]ЃЌОпгаКмИпЕФЛЏбЇКЭЗЧЖдгГбЁдёадЃЈЭМ 21BЃЉЁЃЛњРэбаОПБэУїЧПбѕЛЏжаМфЬх TACЁЄ2+*ПЩДЅЗЂЯЉЬўЕзЮяЕФЕЅЕчзгзЊвЦЩњГЩЯЉЬўздгЩЛљбєРызгЃЌШЛКѓБЛввЫсВЖЛёЃЌНјвЛВНбѕЛЏЮЊбѕЬМжаМфЬхЃЌзюКѓЫЎНтЪЭЗХГіввѕЃбѕЛљєЧЛЏВњЮяЁЃ
2 МИжжГЃМћЗДгІРраЭЕФАБЛљЫсКЭЖрыФЕчЛЏбЇаоЪЮ
2.1 ЭбєШЗДгІ
єШЫсПЩЭЈЙ§бєМЋбѕЛЏзїгУЭбєШаЮГЩЭщЛљздгЩЛљЃЌШнвзгыЫЋМќЗЂЩњМгГЩЗДгІЃЌЛђдк Kolbe ЕчНтЗЈжаНсКЯГЩЖдГЦЕФЖўОлЬхЁЃСэвЛЗНУцЃЌдк Kolbe ЬѕМўЯТаЮГЩЕФздгЩЛљжаМфЬхПЩНјвЛВНбѕЛЏЩњГЩЯргІЕФЬМе§РызгЃЌетвЛЙ§ГЬБЛГЦЮЊЗЧKolbe ЕчНтЛђ Hofer-Moest ЗДгІ[54]ЁЃ2019 ФъЃЌЭѕбЧЛдПЮЬтзщвдМђЕЅЕФ ІС-АБЛљЫсЮЊЕзЮяЃЌЖржжпђРрКЭѕЃАЗРрЮЊ N-ЧзКЫЕзЮяЃЌЭЈЙ§ЕчЛЏбЇбѕЛЏдкЗЧ Kolbe ЬѕМўЯТЪЕЯжСЫ Csp3-N ЭбєШХМСЊЗДгІ[55]ЃЈЭМ 22ЃЉЁЃИУЗНЗЈБмУтСЫєШЫсЕФдЄЛюЛЏЃЌвВЮоашЪЙгУЙ§ЖЩН№ЪєКЭЛЏбЇбѕЛЏМСЃЌЪЧЙтбѕЛЏДпЛЏ ІС-АБЛљЫсЭбєШ C-N ХМСЊЕФвЛжжСМКУВЙГфЁЃ

2020 ФъЃЌЭѕЦНПЮЬтзщЭЈЙ§ЕчЛЏбЇгеЕМЕФ Giese ЗДгІ[56]ЪЕЯжСЫЦпыФЕФЭбєШЛЗЛЏЃЈЭМ 23ЃЉЁЃДјга C ЖЫ N-єЧЛљСкБНЖўМзѕЃбЧАЗбѕЛЏЛЙдЛюадѕЅЃЈRAEЃЉЕФЖрыФдквѕМЋЛЙдЭбєШКѓВњЩњвЛИіЧзКЫЬМжааФздгЩЛљЃЌИУздгЩЛљЛсгыЮЛгк N ЖЫЕФБћЯЉѕЃАЗЗжзгЗЂЩњЗжзгФкЗДгІЁЃСэЭтЃЌЭщЛљ RAE ЕФЕчЛЏбЇЭбєШЗДгІвВБЛгУРДЪЕЯжЭщЛљєШЫсЕФМђЕЅХ№ЫсЛЏ[57]ЁЃзмжЎЃЌетЯюЙЄзїВЙГфСЫЯжгаЕФФјДпЛЏ[58]КЭЙтЛЏбЇ[59]ЖрыФЛЗЛЏЗНЗЈЃЌВЂЮЊНјвЛВНПЊЗЂЕчЛЏбЇЭбєШЖрыФаоЪЮЗНЗЈЕьЖЈСЫЛљДЁЁЃЕЋгЩгкашвЊОРњЭбєШЛЗЛЏЃЌдзгОМУадЛсНЕЕЭЃЌЖјЧвЯрЖдНЯИпЕФгІгУЕчЮЛЃЈ2.5 VЃЉПЩФмгыОпгабѕЛЏЛЙдЛюадЕФАБЛљЫсВрСДЙйФмЭХЃЈШчРвАБЫсЁЂЩЋАБЫсЃЉВЛМцШнЃЌПЩФмЛсЯожЦИУЗНЗЈдкЖрыФКЭЕААзжЪЛЗЛЏжаЕФЙуЗКгІгУЁЃ
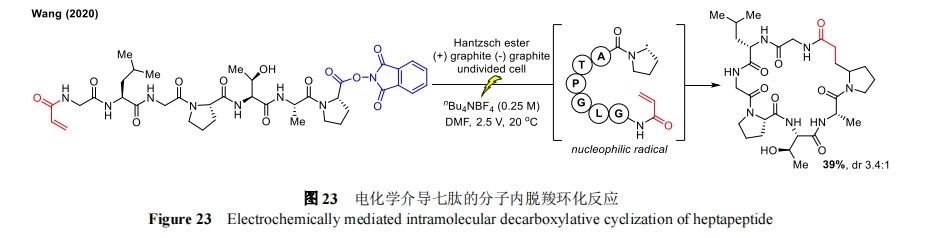
2021 ФъЃЌDavid ПЮЬтзщБЈЕРСЫвЛжжРћгУМфаЊКЭСЌајСїВпТдНјааАБЛљЫсбєМЋЭбєШввѕЃбѕЛљЛЏ[60]ЕФЗДгІЃЈЭМ 24ЃЉЁЃИУЗНЗЈвбгІгУгкЖржжЬьШЛКЭКЯГЩАБЛљЫсЕФбмЩњЛЏЃЌАќРЈКЯГЩЛюадвЉЮяГЩЗжЕФЙиМќжаМфЬхЁЃЮоТлЪЧЕЅДЮЗДгІЛЙЪЧЕчНтвКдйбЛЗЃЌЖМЪЕЯжСЫГіЩЋЕФзЊЛЏТЪКЭбЁдёадЁЃЕЋЪЧетжжЕчНтЗНЗЈВЂВЛЪЪгУгкКЌгабѕЛЏВЛЮШЖЈЙйФмЭХЕФЬьШЛАБЛљЫсЃЌШчАыызАБЫсЕШЃЌЕзЮяЗНУцДцдквЛЖЈЕФОжЯоадЁЃ
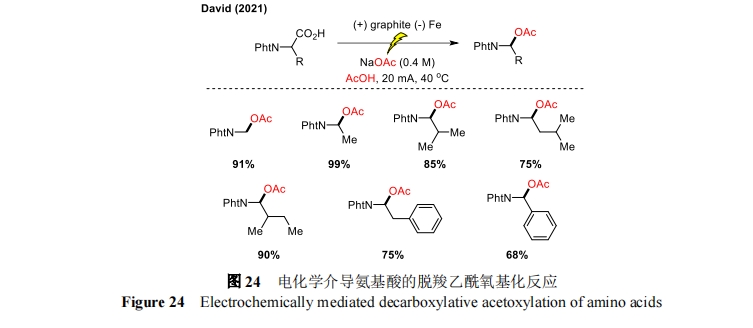
20 ЪРМЭ 80 ФъДњФЉЃЌSeebach ПЮЬтзщЗЂБэСЫПЊДДадЮФеТ[61]ЃЌИХЪіСЫШчКЮРћгУЕчЛЏбЇЗНЗЈдкМзДМШмвКжажБНгбєМЋЭбєШКЯГЩ N ФЉЖЫБЃЛЄЕФЖрыФЁЃЭЈЙ§ЕчЛЏбЇЩњГЩЯргІЕФ N,O-ЫѕШЉЃЌШЛКѓдкЫсадЬѕМўЯТЫЎНтЕУЕНЖрЙйФмЭХЛЏЕФЖрыФВњЮяЃЌЛђЪЧБЛИёЪЯЪдМСЁЂбЧСзЫсбЮЧзКЫНјЙЅЃЌЗжБ№ЩњГЩ C ЖЫЭщЛљ/ЯЉБћЛљЛђСзЫсЖўѕЅЙйФмЛЏыФЁЃетжжКѓЦкаоЪЮЗНЗЈдкКуЖЈЕчСїЬѕМўЯТЕФЮДЗжИюГижаНјааЃЌПЩвдБШНЯШнвзЕиЛёЕУИїжж C ЖЫЙйФмЛЏЕФЖрыФбмЩњЮяЁЃНќЦкЃЌMalins ПЮЬтзщНЋетжжЕчЛЏбЇбѕЛЏЭбєШЗЈгІгУЕНСЫЫЋрѕЃАЗыФЬьШЛВњЮяРрЫЦЮяЕФШЋКЯГЩжа[62]ЁЃИУВпТдРћгУЙиМќЕФЕчЛЏбЇбѕЛЏЭбєШВНжшЩњГЩЛюад N,O-ЫѕШЉЃЌФмЙЛгыИїжжЧзКЫЮяНсКЯЃЌЭЈЙ§ Friede-Crafts ЗДгІЁЂжБНггаЛњН№ЪєМгГЩКЭЛЧѕЃЛЏЛЏбЇЃЌЭъГЩСЫЖржжМЁАБЫсбмЩњЮяЕФЗЂЩЂКЯГЩЃЈЭМ 25ЃЉЁЃетжжЗНЗЈЬсЙЉСЫжЎЧАЮоЗЈЛёЕУЕФ C ЖЫЙІФмЖрыФЃЌБмУтСЫжБНгЛюЛЏЖрыФФЉЖЫЕФШБЯнЃЈШчвьЙЙЛЏЃЉЃЌДгЖјЭЛГіСЫЕчЛЏбЇММЪѕЙЙжўЙІФмЛЏЖрыФЗНУцЕФгХЪЦЁЃ
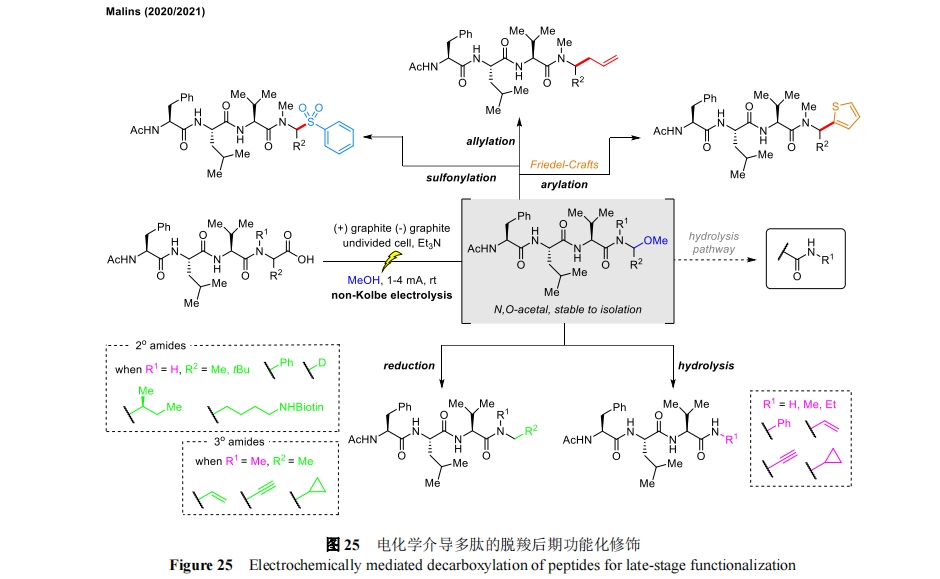
дкетЯюбаОПжаЃЌзїепзЂвтЕН N,O-ЫѕШЉжаМфЬхЕФЫЎНтЪЧвЛИіживЊЕФОКељЭООЖЃЌЬиБ№ЪЧЖдгкЛюадИќЧПЕФ N,O-ЫѕШЉЁЃЫљвдКѓајЕФбаОПжаЭЈЙ§зюГѕЕФЫЎНтЭООЖКЭИќСщЛюЕФЛЙдЭООЖЩшМЦГіСЫ ІС-ѕЃАЗЖрыФ[63]ЁЃИУЗНЗЈПЩЬсЙЉОпгаИпМлжЕЙІФмЛљЭХЕФЖўМЖКЭШ§МЖѕЃАЗЃЌгУгкЭЌЮЛЫиБъМЧКЭЩњЮяХМСЊЗДгІЁЃЭЈЙ§ЙиМќЕФЗЧЖдгГбЁдёадЛЙдЃЌКЯГЩСЫЬьШЛВњЮяЪШЫсадѕЃАЗ A вдМАОпгаЩњЮяЛюадыФЕФЯрЙиРрЫЦЮяЃЌАќРЈПЙАЌзЬВЁЖОЕФЯШЕМыФКЭжЮСЦАЉжЂЕФССБћШ№СжЁЃ
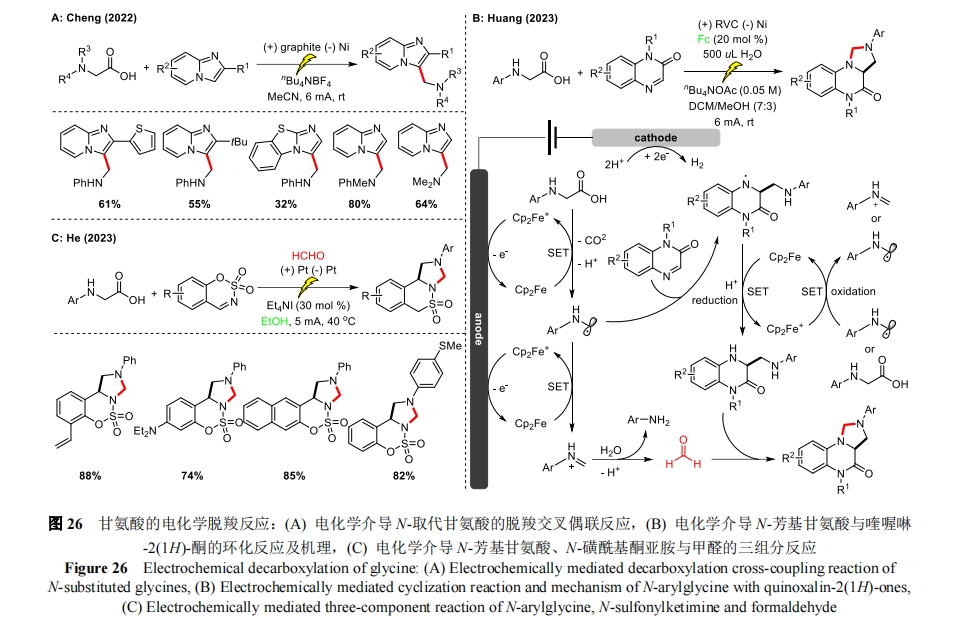
2022 ФъЃЌГЬБѓПЮЬтзщЪЕЯжСЫпфпђдгЛЗгы N-ШЁДњИЪАБЫсЕФЕчЛЏбЇЭбєШНЛВцХМСЊЗДгІ[64]ЃЈЭМ 26AЃЉЁЃЖржж N-ШЁДњИЪАБЫсдкИУЮТКЭЬѕМўЯТОпгаСМКУЕФФЭЪмадЃЌЕУЕНСЫвЛЯЕСа C3 АБМзЛљЛЏпфпђВЂ[1,2-a]пСрЄбмЩњЮяЃЌВЂЪЭЗХГі H2 КЭ CO2ЁЃЛњРэбаОПБэУїжБНггы N ЯрСЌЕФєШЛљбЧМзЛљВПЗжЪЧаЮГЩСйНчздгЩЛљжаМфЬхЕФЙиМќЃЌВЂЧвЭбєШЖдАБЛљбЧМзЛљздгЩЛљЕФаЮГЩвВЪЎЗжживЊЁЃДЫЭтЃЌЛЦОЋУРПЮЬтзщЖдетвЛЭбєШЗДгІНјааСЫНјвЛВНЕФЭиеЙЃЌвдЖўУЏЬњзїЮЊбѕЛЏЛЙдДпЛЏМСЃЌЭЈЙ§ррИпј-2(1H)-ЭЊгы N-ЗМЛљИЪАБЫсЕФЕчЛЏбЇЛЗЛЏКЯГЩЫФЧтпфпђВЂ[1,5-a]ррИпј-4(5H)-ЭЊ[65]ЁЃЦфжаЃЌN-ЗМЛљИЪАБЫсЭЈЙ§ЖўУЏЬњбєРызгЕФЕЅЕчзгзЊвЦЭбєШКѓЩњГЩАБЛљбЧМзЛљздгЩЛљЃЌЫцКѓИУздгЩЛљМгГЩЕНррИпј-2(1H)-ЭЊЩЯЃЌОЖўУЏЬњЛЙдКѓЩњГЩ C3 АБМзЛљжаМфЬхЁЃгыЩЯЪіВЛЭЌЕФЪЧЃЌN-ЗМЛљИЪАБЫсВњЩњЕФАБЛљбЧМзЛљздгЩЛљЛсдкЖўУЏЬњбєРызгЕФзїгУЯТОСэвЛЭООЖбѕЛЏЮЊбЧАЗРызгЃЌдкЫЎЕФДцдкЯТЩњГЩЗМЛљАЗКЭМзШЉЃЌМзШЉЛсНјвЛВНгы C3 АБЛљМзЛљЛЏжаМфЬхЗДгІЩњГЩЛЗЛЏВњЮяЃЈЭМ 26BЃЉЁЃдкДЫЗДгІжаЃЌЖўУЏЬњГ§СЫдкбєМЋбѕЛЏЃЌвВзїЮЊжаМфВњЮяЕФбѕЛЏМСЃЌвђДЫНЕЕЭСЫИУЗДгІЕФКФЕчСПЁЃЭЌФъЃЌКЮЮРУёПЮЬтзщБЈЕРСЫЕчЛЏбЇ EtOH ДпЛЏЕФ N-ЛЧѕЃЛљЭЊбЧАЗЁЂN-ЗМЛљИЪАБЫсКЭМзШЉЕФШ§зщЗжЗДгІ[66]ЃЈЭМ 26CЃЉЁЃЦфжаЃЌEtOH МцОпДпЛЏМСКЭЗДгІШмМСЕФЫЋжизїгУЃЌМђЛЏСЫЗДгІЬхЯЕЁЃ
2.2 ІС-МзбѕЛљЛЏЗДгІ
2023 ФъЃЌChristmann аЁзщвд NaCl ЮЊНщжЪЭЈЙ§ЕчЛЏбЇбѕЛЏЪЕЯжСЫАБЛљЫсЕФ ІС-МзбѕЛљЛЏЗДгІ[67]ЃЌЫцКѓЭЈЙ§ЫсДпЛЏЭбЧтПЩЩњГЩОпгаЧБдкЩњЮяЛюадЕФЭбЧтАБЛљЫсбмЩњЮяЃЈЭМ 27ЃЉЁЃЕБАБЛљБЛ Fmoc БЃЛЄЪБЕзЮяЗжНтЕМжТЗДгІВЛМцШнЃЌПЩФмЪЧдЮЛаЮГЩЕФМзбѕЛљРызггАЯьСЫМюгеЕМЕФЭбБЃЛЄзїгУЁЃЕЋРћгУМюгеЕМЕФМзДМЯћГ§зїгУПЩвдЪЕЯжЭбЧтРЕАБЫсКЭФёАБЫсЕФСЂЬхбЁдёадЛёШЁЁЃ
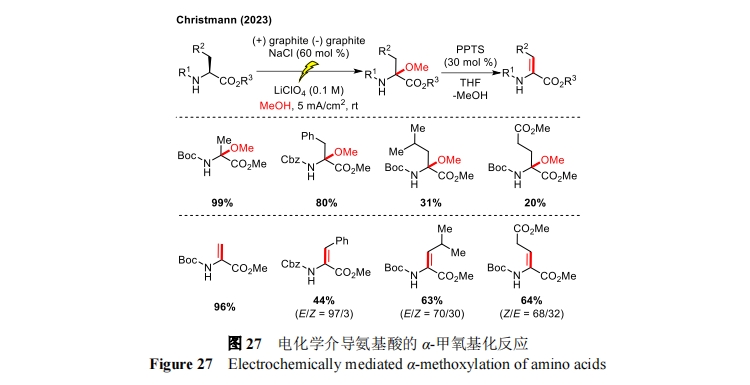
2.3 C ФЉЖЫєШЛљЕФаоЪЮ
2021 ФъЃЌРзАЎЮФПЮЬтзщЪзЯШБЈЕРСЫвЛжжЖўМзЛљСђУбЃЈDMSЃЉНщЕМЕФЛюЛЏАБЛљЫсЕФЕчЛЏбЇбѕЛЏВпТд[68]ЁЃвЛЯЕСаАБЛљЫсПЩвдгыДМЗДгІЃЌЕУЕНСМКУЕФѕЅЛЏВњЮяЃЈЭМ 28ЃЉЁЃЦфжаЃЌФЩИёСаФЮзїЮЊжЮСЦЬЧФђВЁЕФгааЇвЉЮяЃЌвВФмдкИУЗДгІжаЫГРћЕУЕНаоЪЮЁЃЛњРэбаОПБэУїЃЌDMS зїЮЊвЛжжУННщдкАБЛљЫсЕФзЊЛЏЙ§ГЬжаЦ№зХжСЙиживЊЕФзїгУЃЌФмДйНјАБЛљЫсЕФѕЅЛЏЃЌзюКѓвд DMSO ЕФаЮЪНРыШЅЁЃ
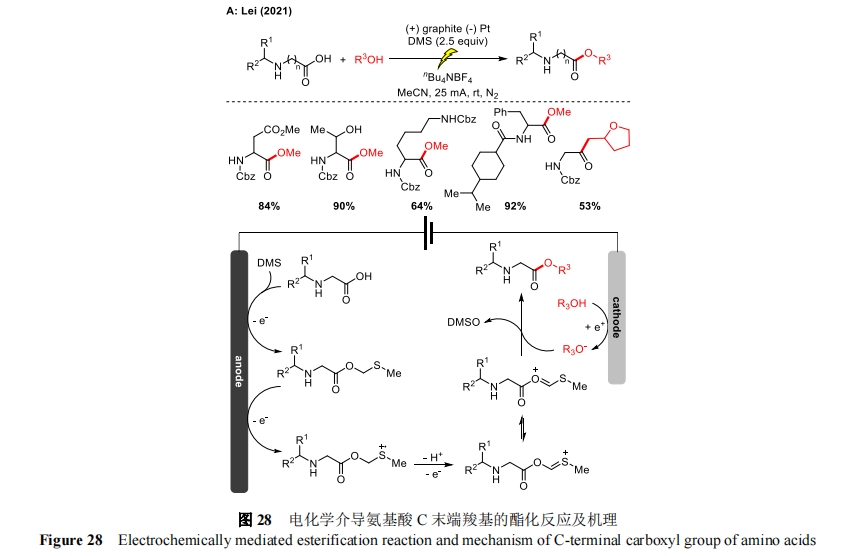
злКЯвдЩЯЗДгІЃЌДгКЯГЩЛЏбЇЕФНЧЖШЗЂЯжЃЌCЈDO МќЕФЙЙНЈЪЧ ІС-ѕЃбѕЛљСђЛЏЮяКЯГЩЕФЙиМќЁЃгыСкНќ O/N дзгЕФ C-H МќЛюЛЏЯрБШЃЌІС-Сђ CЈDH МќЙйФмЛЏОпгаНЯДѓЕФЬєеНадЃЌвђЮЊСђдзгЕФМлЬЌИќЮЊИДдгЁЃдкбѕЛЏМСЕФзїгУЯТЃЌСђЛЏЮязмЪЧКмШнвзБЛбѕЛЏГЩСђУбЛђэП[69]ЁЃР§ШчЃЌРзАЎЮФПЮЬтзщЭЈЙ§бєМЋбѕЛЏгеЕМ ІС Сђ CЈDH МќЛюЛЏЃЌЪЕЯжСЫАБЛљЫсгыСђЛЏЮяЕФНЛВцХМСЊЗДгІ[70]ЃЌКЯГЩГівЛЯЕСа ІС-ввѕЃбѕЛљСђЛЏЮяЃЈЭМ 29AЃЉЁЃЪЕбщЗЂЯжЖржж ІС-АБЛљЫсбмЩњЮяОљПЩдкЪвЮТЯТНјааетжж C-H МќѕЃбѕЛљЛЏЗДгІЁЃДЫЭтЃЌІТ-КЭ ІУ-АБЛљЫсвВФмвдСМКУЕФЪеТЪЕУЕНЯргІЕФВњЮяЁЃНЋКЌМзЛљСђЛЏЮягыАБЛљЫсЦЌЖЮНсКЯКѓПЩвдгыКЌєШЫсЕФвЉЮяЗжзгСЌНгЃЌєШЫсЛљЭХКЭМзЛљСђУбГЩЙІЕизїЮЊФПБъЛљЭХзМШЗЕиЖдЖрыФКЭвЉЮяЗжзгНјааЮЛЕубЁдёадКЯГЩЃЌШчВМТхЗвКЭБћЛЧЪцЁЃИќживЊЕФЪЧЃЌИУЗДгІЪЪгУЖржжВЛЭЌЕФЧзКЫЕзЮяЃЌШчБНВЂШ§пђбмЩњЮяЃЈЕЊдгЛЏЃЉЁЂДМЃЈУбЛЏЃЉКЭввЫсяЇЃЈѕЃбѕЛљЛЏЃЉЁЃжЕЕУвЛЬсЕФЪЧЃЌвЛжжЪмБЃЛЄЕФЕААБЫсбмЩњЮявВФмгыБНВЂШ§пђбмЩњЮяЫГРћНјаа ІС-СђЕЊдгЛЏЩњГЩЯргІВњЮяЁЃЦфжаЃЌдіДѓЕчСїЃЈДг 11 mA ЕН 25 mAЃЉДйНјСЫКЌЕААБЫсЖўыФЕФѕЃбѕЛљЛЏЁЃетжжзЊЛЏЫфШЛжЛЪЧЕЅИіАБЛљЫсКЭЖўыФЃЌЕЋШДДцдкзХЬиЪтвтвхЃЌвђЮЊЦфРћгУСЫдЩњЕААзжЪЕФЙІФмЃЌЭЛГіСЫЕААБЫсСђУбВрСДНјааЩњЮяЙВщюЕФЕчЛЏбЇЧБСІЁЃЛњРэбаОПБэУїздзщзАгеЕМЕФ C(sp3)-H/O-H ёюКЯЭООЖОпгааЭЌаЇгІЃЌMeOH ЕФМгШыЪЧИУЗНАИОпгаЧјгђбЁдёадКЭЙуЗКЪЪгУадЕФЙиМќЫљдкЁЃСђЛЏЮяЁЂєШЫсКЭ MeOH зщзАГЩМгКЯЮяЃЌєШЫсКЭСђдзгжЎМфЕФЧтМќПЩДйНјСђЛЏЮяЕФ SET бѕЛЏЁЃВЛНіШчДЫЃЌMeOH ПЩбЁдёадЕиВЖЛёжЪзгЃЌаЮГЩОпгаИпЧјгђбЁдёадЕФздгЩЛљжаМфЬхЃЌНгзХЃЌБЛбєМЋбѕЛЏЪЇШЅвЛИіЕчзгЩњГЩСђе§РызгжаМфЬхЃЌзюКѓБЛєШЫсЧзКЫЙЅЛїЕУЕНЫљашЕФВњЮяЃЈЭМ 29BЃЉЁЃ
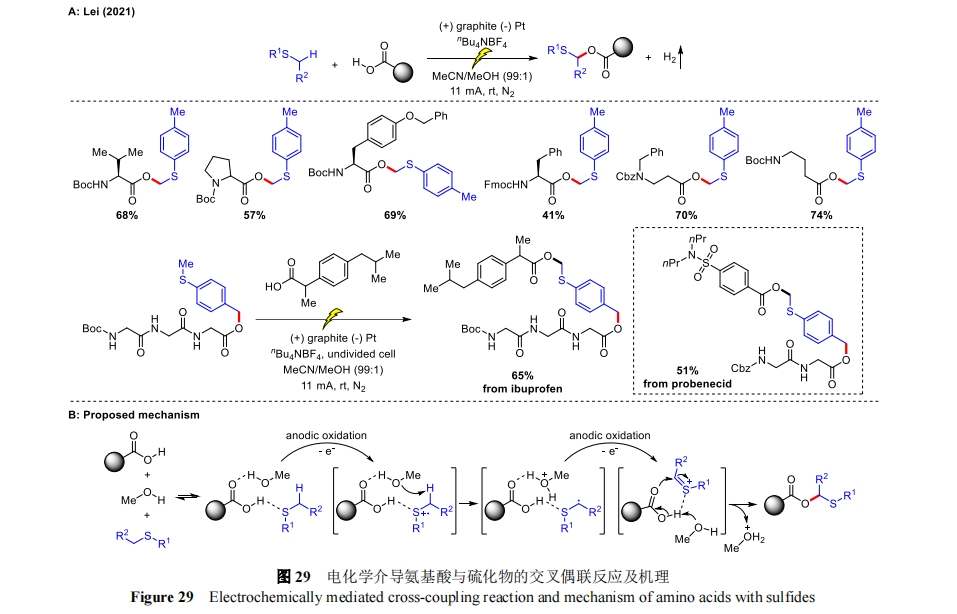
3 ЕчЛЏбЇЖрыФКЯГЩ
Г§СЫАБЛљЫсВаЛљгыЖрыФВрСДЕФаоЪЮЭтЃЌаЁЗжзгЪдМСЕФМфНгЕчЛЏбЇЛюЛЏПЊЗЂГіСЫвЛжжыФХМСЊЗНАИЃЌгыДЋЭГЗНЗЈЯрБШЃЌИУЗНАИЬсИпСЫдзгОМУадЃЌМѕЩйСЫРЫЗбЁЃдкЯШЧАБЈЕРЕФЕчЛЏбЇѕЃАЗМќЙЙНЈ[71]ЕФЛљДЁЩЯЃЌChiba НЬЪкНЋЕчЛЏбЇгыаТаЭЕФПЩШмаджЇГжЮяИЈжњВпТдНсКЯЦ№РДЃЌДйНјСЫѕЃАЗЙЧМмЕФИпбЁдёадКЯГЩ[72]ЃЌВЂЧвВЛЛсЗЂЩњЯджјЕФВюЯђвьЙЙЛЏЁЃИУЗНЗЈРћгУШ§БНЛљьЂЕФбєМЋбѕЛЏзїгУЩњГЩьЂздгЩЛљбєРызгЃЌИУбєРызгБЛєШЫсХМСЊМСВЖЛёЃЌаЮГЩЛюЛЏЕФѕЃбѕЛљШ§БНЛљьЂРызгЁЃАЗХМСЊЮяЫцКѓНјааЧзКЫНјЙЅЭъГЩЫѕКЯЗДгІЃЌВЂЪЭЗХГіШ§БНЛљбѕьЂЃЈTPPOЃЉзїЮЊЛЏбЇМЦСПИБВњЮяЃЈЭМ 30CЃЉЁЃживЊЕФЪЧЃЌЫљгаГЃМћЕФЕААзАБЛљЫсЃЌЮоТлЪЧзїЮЊ Fmoc БЃЛЄЕФєШЫсХМСЊЮяЃЌЛЙЪЧзїЮЊ C ЖЫБъМЧЕФАЗХМСЊЮяЖМгыИУЗНАИМцШнЁЃжЕЕУзЂвтЕФЪЧЃЌЕБ Fmoc-His(Trt)-OH ХМСЊЪБЙлВьЕНСЫВПЗжвьЙЙЛЏЯжЯѓЃЌвд Boc ЬцДњ Fmoc зїЮЊзщАБЫсВрСДЕФБЃЛЄЛљЭХКѓПЩвдНтОіетвЛЮЪЬтЃЈЭМ 30AЃЉЁЃГ§СЫЖўыФФЃПщЭтЃЌChiba ЛЙКЯГЩСЫССБћШ№СжЃЈLeuprorelinЃЉЃЌЫќЪЧвЛжжКЌ8 ИіВаЛљЕФЖрыФЃЌзїЮЊЛюадвЉЮяГЩЗжОпгаживЊЕФЩЬвЕМлжЕЃЈЭМ 30BЃЉЁЃзюКѓЃЌУПвЛВН Fmoc ЕФЭбБЃЛЄКЭЕчЛЏбЇыФХМСЊЕФЦНОљЪеТЪЖМДѓгк 95%ЃЌТњзуСЫЙЬЯрыФКЯГЩЃЈSPPSЃЉЛЏбЇЫљвЊЧѓЕФЕќДњЭбБЃЛЄКЭХМСЊбЛЗЕФаЇТЪБъзМЁЃ
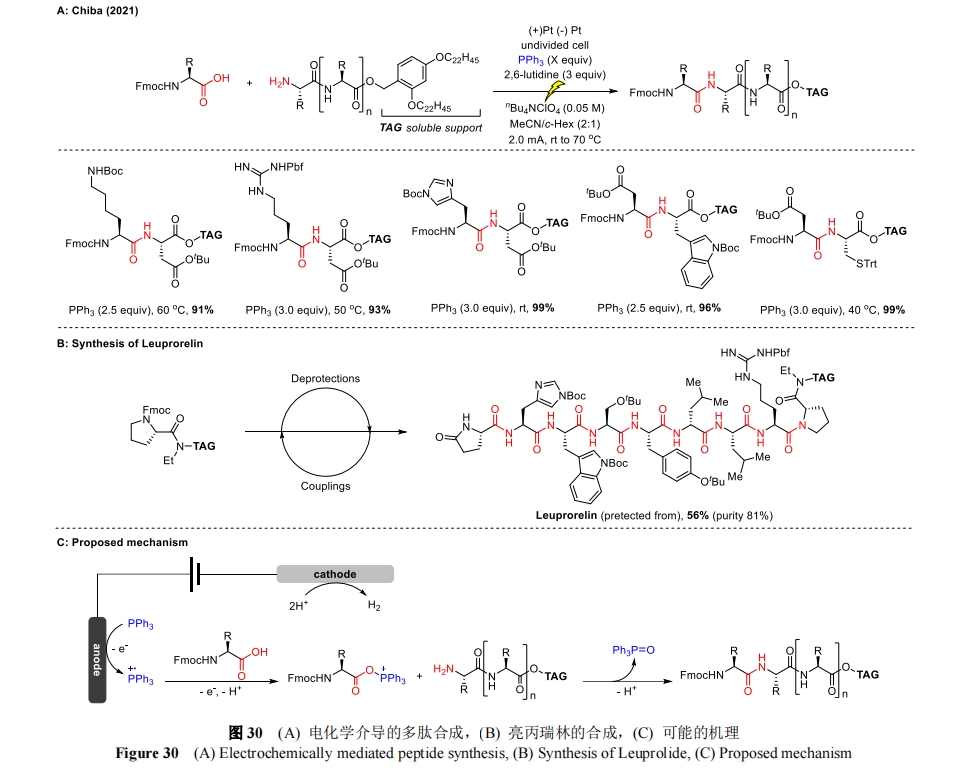
ЩЯЪіЗНЗЈжЎЫљвдФмЙЛШЁЕУГЩЙІЃЌЪЧвђЮЊЪЙгУСЫвЛжжЖРЬиЕФЫЋЯрЯЕЭГЃЌИУЯЕЭГгЩввыцКЭЛЗМКЭщ[73]вдМАМЋЪшЫЎЕФ CЖЫБъЧЉзщГЩЁЃетжжЩшМЦПЩвдЪЙШ§БНЛљьЂКЭєШЫсЃЈдкввыцжаЃЉгыАЗХМСЊЮяКЭбгГЄЕФыФСДЃЈдкЛЗМКЭщжаЃЉЗжРыЃЌДгЖјЭЦЖЏЕчЛЏбЇЗДгІЕФЭъГЩЃЌВЂЗРжЙАЗЦ№ЪМЮяКЭХМСЊВњЮяЗЂЩњИБЗДгІЁЃДЫЭтЃЌИУЗНЗЈЛЙФмЗНБуЕиДг TPPO ИБВњЮяжаЗжРыКЭЛиЪеЫљашЕФыФЁЃвђДЫЃЌетЯюММЪѕЭЙЯдСЫЕчЛЏбЇЗНЗЈЕФПЩГжајадКЭЖрЙІФмадЃЌЮЊШчКЮРћгУЕчЛЏбЇРДжиаТЩшМЦЯжгаЕФЗДгІЃЈШчєШЫсЕФЛюЛЏЃЉЬсЙЉСЫвЛИіКмКУЕФР§згЃЌВЂНтОіСЫЖрыФКЯГЩжаГЄЦкДцдкЕФФбЬтЁЃПМТЧРћгУЕчЛЏбЇММЪѕдкДПЫЎНщжЪжаИпаЇКЯГЩЖрыФвВКмгавтвхЃЌОЁЙмзюНќШЁЕУСЫвЛаЉНјеЙ[74]ЃЌЕЋетШдШЛЪЧИУСьгђЕФвЛЯюжиДѓЬєеНЃЌЬиБ№ЪЧЖдгкГЄЖШГЌЙ§ 10 ИіАБЛљЫсВаЛљЕФЖрыФЖјбдЁЃ
4 змНсгыеЙЭћ
БОЮФзмНсСЫНќЮхФъРДЕчЛЏбЇаоЪЮАБЛљЫсКЭЖрыФРрЛЏКЯЮяЕФЗНЗЈЃЌИљОнАБЛљЫсжжРрМАЯрЙиЗДгІРраЭНјааСЫЗжРрЁЃетаЉЗНЗЈЗДгІЬѕМўЮТКЭЃЌОпгаЙЬгаЕФПЩЕїадЃЌПЩвдбЁдёдЩњЙІФмРДаоЪЮАБЛљЫсКЭЖрыФЕзЮяЃЌзюДѓЯоЖШЕФБЃСєСЫАБЛљЫсВрСДЕФМцШнадЁЃЕЋЪЧФПЧААБЛљЫсЕФЕчЛЏбЇаоЪЮжївЊМЏжадкРвАБЫсКЭЩЋАБЫсВаЛљЩЯЃЌЦфЫћАБЛљЫсВаЛљЕФаоЪЮНігаЩйСПБЈЕРЁЃвЛЗНУцЃЌЗДгІРраЭжївЊМЏжагкЭбєШЗДгІЃЌЭЈЙ§ЕчЛЏбЇЪЕЯжЖрыФКЯГЩЕФбаОПНігавЛР§ЃЌКѓајПЩЭЈЙ§ЕїНкЗДгІЬѕМўЃЌЩшМЦЖрыФжаИјЖЈВаЛљжмЮЇЕФЮЂЛЗОГЃЌЕїећЦфбѕЛЏЛЙдЕчЮЛвдЪЕЯжЦфЫћАБЛљЫсЕФЕчЛЏбЇаоЪЮЃЌЗсИЛЗДгІРраЭЁЃСэвЛЗНУцЃЌФПЧАЖдгкАБЛљЫсКЭЖрыФЕФСЂЬхбЁдёадЃЈВЛЖдГЦЃЉаоЪЮвВЯЪгаБЈЕРЃЌЮДРДПЩПМТЧдкЗДгІжаМгШыЪжадХфЬхЛђЩшМЦКЌгаЪжаджааФЕФЕзЮявдгеЕМЪЕЯжВЛЖдГЦКЯГЩЃЌгЩДЫзїЮЊЖрыФаоЪЮЕФвЛИіЗЂеЙЗНЯђЁЃжЕЕУзЂвтЕФЪЧЃЌЦљНёЮЊжЙЕФДѓЖрЪ§баОПЙЄзїЖМЪЧдкгаЛњШмМСКЭЮДЗжИюЕФЕчНтГижаЪЙгУКуСїЕчНтЗЈНјааЕФЁЃвђДЫЃЌКѓајашвЊЬНЫїЫЎадШмМСгыАБЛљЫсВрСДМцШнЕФаТаЭаоЪЮММЪѕЃЌвдШЗБЃзюДѓЯоЖШЕиЪЪгУгкИїжжЕзЮяЁЃДЫЭтЃЌвВгІЬНЫїЗжИюЕчНтГигыКубЙЕчНтЕФгІгУЃЌЫфШЛдкВйзїЩЯЛсИќЮЊИДдгЃЌЕЋетаЉЗДгІЩшжУПЩвдЬсЙЉИќКУЕФПижЦКЭПЩЕїадЃЌДгЖјЬсИпЛЏбЇбЁдёадЁЃСэЭтЃЌЯжгаЗНЗЈжаЪЙгУЕФЕчМЋХфжУЗЖЮЇКмЙуЃЌетЭЙЯдСЫЩИбЁИїжжЕчМЋВФСЯЖдгкЬНОПЗДгІбЁдёадКЭзюДѓЛЏВњТЪЕФживЊадЁЃвЛАуЕиЃЌЬМЕчМЋКЭВЌЕчМЋгЩгкЦфЮШЖЈадКЭЖшадЃЌвдМАЃЈЬМЕчМЋЃЉЯрЖдНЯЕЭЕФГЩБОЃЌПЩФмЛсМЬајзїЮЊзюМбЕчМЋбЁдёЁЃзюКѓЃЌЯЃЭћРћгУЕчЛЏбЇВпТдЕФЮТКЭадЁЂЁАЮоВаСєЁБадКЭНчУцадФмЮЊбЁдёадаоЪЮАБЛљЫсКЭЖрыФЬсЙЉЖРЬиЕФЗНЗЈКЭЙтУїЕФЧАОАЁЃ
Утд№ЩљУїЃКБОЮФЮЊаавЕНЛСїбЇЯАЃЌАцШЈЙщдзїепМАддгжОЫљгаЃЌШчгаЧжШЈЃЌПЩСЊЯЕЩОГ§ЁЃЮФеТБъзЂгазїепМАЮФеТГіДІЃЌШчашдФЖСдЮФМАВЮПМЮФЯзЃЌПЩдФЖСддгжОЁЃ
