
摘 要 多肽由于具有良好的生物相容性和生物可降解性、生物活性以及自组装特性, 近年来受到了广泛的关注。将多肽自组装特性引入到聚合物中,可赋予聚合物形成凝胶性并对凝胶网络分子结构做出一定控制,进而使凝胶具有如环境响应、力学可调等结构控制性能;将特殊功能性多肽引入到化学交联的聚合物凝胶网络中,可赋予水凝胶生物功能性,如细胞黏附、酶降解、抗菌等;将多肽的凝胶网络构建、结构控制作用以及功能性同时引入获得的物理/ 化学双重交联凝胶不仅赋予水凝胶一定的功能性,且多肽自组装贡献的物理交联结构还能对化学交联凝胶网络起增强作用。本文综述了基于多肽自组装的物理交联聚合物水凝胶、多肽功能化的化学交联聚合物水凝胶以及基于多肽的物理/ 化学双重交联的聚合物水凝胶,并展望了这些水凝胶的发展前景。
水凝胶是由三维交联网络结构和介质水共同组成的多元体系,其交联网络中含有大量的亲水性基团,当水分子扩散到凝胶网络中时,水凝胶吸水后体积可膨胀至原来数倍。在过去几十年里,水凝胶由于具有含水量高、结构与细胞外基质相似以及生物相容性良好等特点,其在药物释放[1]、组织工程[2]、细胞培养[3]以及生物黏合[4,5] 等医用领域得到了广泛应用。
自然界存在的天然氨基酸仅有 20 种,一些非天然氨基酸也可实验合成得到,它们具有不同的物化性质:极性、酸碱性以及是否含芳香基团。有限种类的氨基酸通过肽键按一定序列排列可得到大量具有不同理化性质的多肽[6]。由于多肽链段上含有大量肽键以及不同结构的氨基酸残基,使其可利用彼此间的氢键等非共价键作用来实现分子的有效自组装,比如卷曲螺旋结构、β⁃折叠结构、三股螺旋结构以及更高级结构。将多肽自组装特性引入到聚合物中,使得在纳米水平上有序控制其分子结构成为可能[7,8],同时也赋予聚合物凝胶以优良特性,比如外界刺激响应性、力学可调性。多肽还有其他特殊功能性:细胞黏附性、生物可降解性、抗菌性等,比如含精氨酸⁃甘氨酸⁃天冬氨酸(Arg⁃Gly⁃Asp,RGD) 序列的多肽能改善水凝胶的细胞黏附性[9]。本文根据多肽在凝胶体系中的不同作用针对近期基于多肽结构的各类水凝胶进行了综述,并对研究过程中出现的问题作了简要分析。
2 物理交联水凝胶
水凝胶基于交联键性质不同,分为化学或物理凝胶。化学水凝胶是一种永久性凝胶, 即凝胶过程不可逆,而物理凝胶可通过非共价键相互作用等物理方法交联得到,其凝胶过程具有可逆性。多肽可利用静电作用、疏水作用、π⁃π 堆积以及氢键作用等自组装成不同的二级或高级结构,比如卷曲螺旋(coiled coil)、 β⁃折叠 ( β⁃sheet)、 三 股 螺 旋 ( triplehelix),且组装过程具有可逆性[8]。将这些可物理组装的结构引入到聚合物体系中,就会使聚合物形成物理凝胶网络结构,且多肽组装的本质特性还会同时赋予水凝胶以环境响应、力学调控等多种结构控制性能。
不管在自然界还是在实验室中,卷曲螺旋的超二级结构形成都是通过两个或多个 α⁃螺旋多肽分子在水溶液以特定方式捆绑来完成的,这种捆绑特性主要 来 自 α⁃螺 旋 多 肽 分 子 的 七 肽 重 复 序 列(abcdefg)n 结构,其多肽分子重复序列中的 a、d、e、g 位置的氨基酸残基之间的相互作用是构建卷曲螺旋的内在作用力,其中位置 a 和 d 主要由疏水的氨基酸残基所占据,分子间主要为疏水性作用力,而卷曲螺旋结构内部的疏水性强弱主要由 e 和 g 位置的氨基酸残基之间的静电作用来调节,对 α⁃螺旋的稳定性具有重要的作用。
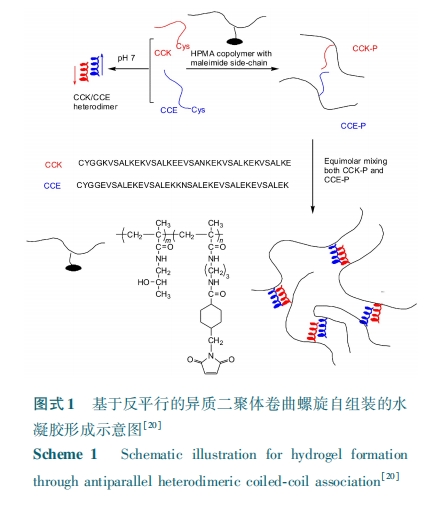
早在 1998 年,Petka 等[10] 就利用 DNA 重组技术设计了一种三嵌段共聚物:共聚物端头为亮氨酸拉链结构,中间为水溶性、分子链无规卷曲的聚电解质。其中亮氨酸拉链结构域可形成卷曲螺旋聚集域而促使聚合物三维网络结构形成,得到了具有外界响应性的水凝胶。利用该设计方法,Xu 等[11] 也设计了一系列的三嵌段共聚物,只替换了端头的亮氨酸拉链结构,中间链段仍为亲水的聚电解质区域,得到的水凝胶同样能够响应外界环境的刺激,且两端头卷曲螺旋域的氨基酸序列对水凝胶的结构和性质有着重要影响。还有研究者通过控制此类分子的网络拓扑结构来达到调节该水凝胶侵蚀率的目的,主要在于压制了环状分子形成[12]。以上的体系都不含人工合成聚合物,为了将 α⁃螺旋多肽和聚合物两者优良性能结合在一起,有研究者将卷曲结构引入到聚乙二醇(PEG)分子中,发现卷曲螺旋的自组装能力依然存在,且通过控制氨基酸的序列可控制此类共聚物的结构和性能[13 ~ 15]。以上的卷曲螺旋主要来自非人类细胞内的多肽,为了减少此类材料用于体内时所引起的免疫原性,Jing 等[16] 对来源于人纤维蛋白螺旋区的多肽序列中特定位置氨基酸残基作了替换,并将其接枝到了 PEG 分子的两端,得到的三嵌段共聚物在多肽形成二聚体或四聚体作用下得到了黏弹性的物理凝胶。
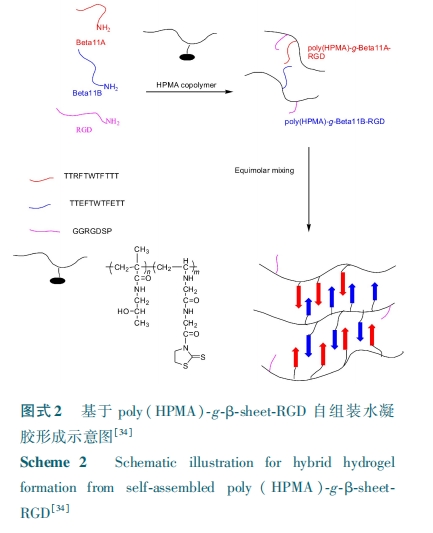
透明质酸有着广泛的应用,可以调节其流变学性质来满足各种应用,Elder 等[36] 将 LSLSLSLS 多肽接枝在透明质酸(HA)的主链上,形成的多级 β⁃折叠结构在 HA 分子链间起到了物理交联作用,和没有多肽修饰的 HA 相比,β⁃折叠明显改善了 HA 的低剪切黏度。
也有研究者将 β⁃折叠结构引入到具有温敏性的聚 N⁃异 丙 基 丙 烯 酰 胺 ( PNIPAAm ) 的 体 系中[37 ~ 39],Miller 课 题 组 最 开 始 将 端 头 巯 基 化 的FEFEFKFK 八肽作为自由基聚合的 PNIPAAm 的链转移剂,将这种八肽接枝到了 PNIPAAm 上,得到了具有双温度响应性的水凝胶,这为合成多肽和温敏性聚合物的新型温敏性大分子开辟了新的途径。纯PNIPAAm 不能形成稳定的物理凝胶,为了避免化学交联剂对组织的毒副作用,该课题组通过链转移反应 将 不 同 含 量 的 PNIPAAm⁃FEFEFKFK 引 入 到PNIPAAm 中,调节两者比例来改变其体系中的自组装程度,进而控制凝胶网络中的物理交联密度,从而得到了力学性能可调的水凝胶。此外,为了赋予没有最低临界溶液温度的 FEFEFKFK 水凝胶以温敏性, 该 课 题 组 又 将 PNIPAAm⁃FEFEFKFK 加 入 到FEFEFKFK 的 体 系 中, 研 究 发 现 PNIPAAm 随 着PNIPAAm⁃FEFEFKFK 和 FEFEFKFK 自组装被固定在纳米纤维结构上,但这并没有影响 PNIPAAm 的最低临界溶液温度,且 PNIPAAm⁃FEFEFKFK 的加入明显增加了水凝胶的剪切模量。相比于单一温敏性凝胶,这类力学性能可调的温敏性凝胶有着更广阔的应用前景。
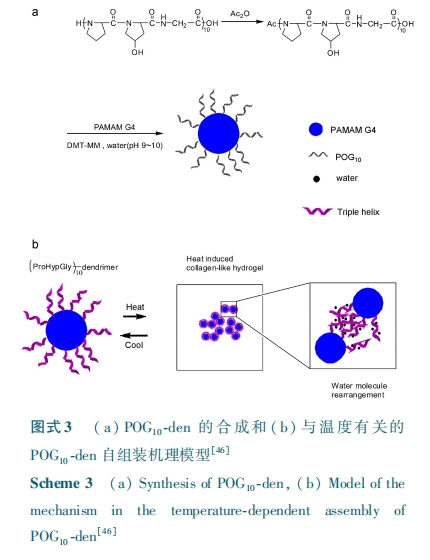
胶原模拟多肽( collagen mimetic peptide,CMP)通常由 15 ~ 45 个氨基酸残基构成,其中重复三肽(Gly⁃Xaa⁃Yaa)n 的存在使其具有自发形成三股螺旋结构的特性,其中 Xaa 通常为脯氨酸( Pro),Yaa 通常为羟脯氨酸(Hyp)。不同于天然的胶原,三股螺旋解链行为具有温度可逆性,且其解链温度可通过多肽的链长和氨基酸组成来改变。过去十几年,随着固相多肽合成技术的发展,在实验室就可利用成熟的技术合成具有特定长度和特定序列胶原模拟多肽。一些典型胶原模拟多肽,比如(Gly⁃Pro⁃Hyp)10(POG10 ) 和 ( Gly⁃Pro⁃Pro)10 ( PPG10 ) 也 已 商 业 化。CMP 的三股螺旋结构还可自组装成更高级的结构,Brodsky 课题组曾经报道过胶原模拟多肽 POG10 可组装成枝状的细丝结构[40]。
单独的 CMP 很难形成热稳定的三股螺旋结构。为了解决这一问题,各种共价键结被用于稳定 CMP三股螺旋结构[41],比如半胱氨酸结、三官能度有机物。树枝状高分子( dendrimer) 外围表面含有高密度基团,使其也可作为稳定 CMP 三股螺旋结构的共价键结。Kinberger 等[42,43] 将树枝状分子作为稳定三股螺旋结构的共价键结,研究发现(Gly⁃Pro⁃Nleu(N⁃isobutylglycine))修饰的树枝状分子中形成了类胶原蛋白的三股螺旋结构,这为进一步利用 CMP 自组装性来形成树枝状水凝胶提供了研究基础。基于三股螺旋结构的温度可逆性以及其自组装性得到的水凝胶,其中多肽的长度和序列组成对最后大分子的结构有着重要的影响。Kojima 等[44] 将(Gly⁃Pro⁃Pro)5 ( PPG5 ) 接到四代聚酰胺⁃胺型树枝状高分子(PAMAM)外围表面得到了 PPG5⁃den,由于很大数量的三股螺旋结构处于低稳定状态,使得体系不能形成足够的物理交联网络结构,它只能在乙醇和硫酸钠的存在下才可形成水凝胶。肽链长度对三股螺旋的稳定性有着重要的影响,因此可增加肽链长度来提高三股螺旋结构稳定性,从而在没有任何添加剂的情况下就可使其组装成水凝胶。Suehiro 等[45]将(Gly⁃Pro⁃Pro)10接到四代 PAMAM 的表面,得到的PPG10⁃den 在没有任何试剂作用下就可形成水凝胶,这得益于 PPG10在 PAMAM 表面的团簇效应以及肽链长度的增加,但其凝胶行为类似于明胶。
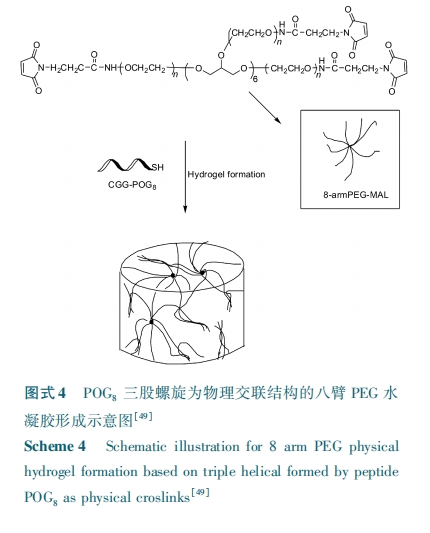
上述研究都是集中于三股螺旋的温敏性和自组装性,而没有研究多臂 PEG 的臂数对形成大分子的构象和交联性质的影响。Matsusaki 等[51] 将 POG10修饰到了多臂 PEG 的端头上(n arm PEG, n = 1,2,4,8)得到了一系列的超分子结构,通过研究发现 narm PEG⁃POG10的流体直径随着 n 的增多先增大后减小,其中 4 arm PEG⁃POG10具有最大流体直径,且温度对流体直径也有一定的影响。由于三股螺旋间可通过氢键相互作用,他们将 POG10 修饰的多臂PEG 作为Ⅰ型胶原的纳米凝胶因子。通过研究发现 4 arm PEG⁃POG10交联的胶原凝胶具有最大的储能模量,从而可推断 PEG 的臂数确实影响到了其体系中三股螺旋的密度,进而影响了胶原水凝胶体系中的物理交联密度。研究也发现 4 arm PEG⁃POG10的浓度亦对其交联的胶原凝胶的储能模量有较大的影响,该凝胶的最大储能模量可达 2.5 kPa。
以上基于 β⁃折叠结构、卷曲螺旋结构以及三股螺旋结构自组装得到的水凝胶具有很多优异的结构控制特性,比如外界环境刺激响应性。同时大部分该类聚合物在由溶液到凝胶转变的过程中都需经过一定的时间,这为手术操作提供了便利,因此未来该类凝胶可望通过调节凝胶制备参数从而通过原位注射的方式在缺损部位形成所需的任意形状来填补缺损或者黏合受损的组织。然而,该类水凝胶网络基本都是物理交联所得,力学性能较差,其凝胶时间及凝胶性质具有明显的浓度以及保温时间依赖性,凝胶的外界刺激响应能力也有待进一步提高,这些也在一定程度上阻碍了其在生物医用领域的应用。多肽的存在使得这类物理凝胶在体内易蛋白水解,尽管人工合成聚合物本身一定程度上能缓解多肽蛋白水解的问题,但这方面研究工作仍面临着诸多的挑战。
3 化学交联水凝胶
多肽自组装主导了上述聚合物物理凝胶的形成,并提供了凝胶以优良的结构控制特性。然而一般来讲物理凝胶的力学性能大多较弱,所以众多研究将焦点关注到化学键交联的水凝胶上。同时值得注意的是在上述物理凝胶中,某些多肽已起到了除结构控制之外的特殊功能化作用,比如基于 β⁃折叠的特种多肽组装形成的纳米纤维可促进羟基磷灰石矿化[34]。因此,现今一些报道将多肽引入到化学交联的凝胶网络中,这里的多肽主要作用不在于其对凝胶结构形成的贡献,而是为了赋予原本的化学凝胶以生物学功能,比如细胞黏附、酶降解、抗菌性等。
不同性能的人工合成聚合物大多具有足够的力学稳定性、弹性、无毒以及抗降解稳定性,被广泛应用于医用方面,但其和细胞之间的作用力不足,易导致体内发炎、血栓形成等不良反应。为了解决这一问题,最初主要使用黏附性的蛋白来修饰材料的表面来改善材料对细胞的黏附性,但这样的方法存在诸多缺点,比如易蛋白降解、免疫原性[52]。在后来研究中发现这些黏附性蛋白的细胞黏附位点仅由数个氨基酸组成,其中研究比较深入、应用比较广泛的是含 RGD 序列多肽,由于这种三肽能与细胞表面的整联蛋白受体结合,从而可促进细胞在材料表面的吸附。含 RGD 序列的多肽包括两种,含 RGD 序列的线性多肽和含 RGD 序列的环形多肽,下面仅对含RGD 的线性多肽作简要的总结.
在过去的研究中,为了改善细胞在水凝胶网络中的黏附、扩散以及增殖性,含 RGD 序列多肽已被引入到透明质酸[53 ,54]、 聚乙烯醇 ( PVA)[55] 以及PEG[56 ~ 59]等水凝胶的体系中。引入的方法主要有两种:一是 RGD 随着交联剂引入到化学网络结构中,比如 Shu 等[53]利用 CCRGDS 多肽序列中双巯基和聚乙二醇二丙烯酸酯(PEGDA)的迈克尔加成反应,得到了分子端头含有双键,中间含有 RGD 的PEGDA⁃RGD⁃PEGDA 交联剂,再和巯基修饰透明质酸间的迈克尔加成反应将 RGD 引入到水凝胶的化学网络中;二是先将 RGD 作为支链部分接枝到聚合物的主链上,然后 RGD 随着聚合物的交联被引入到化学网络中,比如 Schmedlen 等[55] 分别将双键基团和 RGD 通过化学修饰嫁接到了 PVA 的侧链上,最后光交联得到了具有细胞黏附性的水凝胶。除此之外,水凝胶三维网络体系中 RGD 对细胞的增殖以及组织的生成影响具有浓度依赖性[60]。
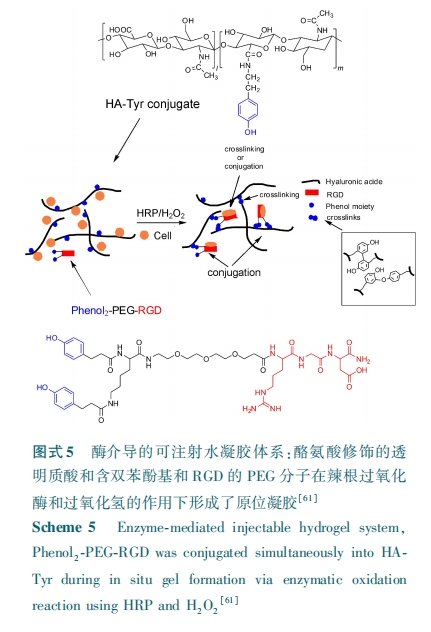
支架材料降解速率是植入材料需要考虑的最重要问题之一,希望其降解速率与组织缺陷位置新组织再生的速率相匹配,如果其降解速率相对于组织再生速率过快,就失去了它对细胞生长的载体功能。反之,就会阻碍新组织的再生。而大多数可降解合成聚合物,比如聚己内酯、聚乳酸、聚乙交酯的降解主要是基于主链酯的随机水解,这些水解过程对细胞信号以及细胞分泌的酶并不具有响应性。为了赋予支架材料生物降解性,其中最好的方法就是利用细胞外基质蛋白水解降解机制,将对酶具有响应性的多肽序列引入到支架材料中[62]。
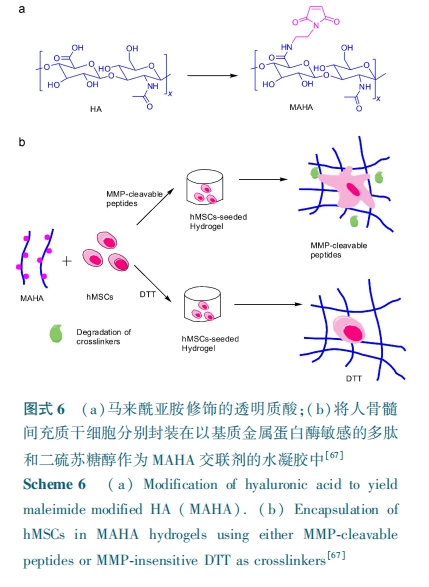
最近几年有研究者报道了将多肽的抗菌性引入到化学交联的聚合物中,从而赋予水凝胶以抗菌性。Zhou 等[70]通过 ε⁃聚赖氨酸(EPL)端头氨基和甲基丙烯酸(MA)羧基间的酰胺化反应得到了 EPL⁃MA,然后将其和 PEGDA 以及 N, N⁃二甲基丙烯酰胺三者在光交联作用下得到了水凝胶,由于阳离子的EPL 可吸附在生物膜的表面导致细胞的生理损伤,使得凝胶具有广谱的抗菌性。Song 等[71] 合成了一系列的 poly( Lys)x (Ala)y ( x + y = 100, Lys:赖氨酸,Ala:丙氨酸), 其结构中赖氨酸残基不仅可作为六臂聚乙二醇酰胺基琥珀酰亚胺碳酸酯 ( 6 armPEG⁃ASG) 的交联剂,同时赖氨酸残基的存在也赋予了水 凝 胶 以 抗 菌 性。进 一 步 研 究 发 现 poly(Lys)60 (Ala)40 和6 arm PEG⁃ASG 交联得到的水凝胶同时具有抗菌性和细胞黏附性,使其可作为潜在的皮肤创伤愈合支架材料。除了聚赖氨酸外,还有研究者将其他具有抗菌性的多肽引入到聚合物中,Cleophas 等[72 ,73] 将 含 有 半 胱 氨 酸 的 抗 菌 肽(CKRWWKWIRW)、季戊四醇四⁃3⁃巯基丙酸酯以及PEGDA 三者的混合液转移到聚对苯二甲酸乙二醇酯片表面,然后通过硫醇⁃烯光聚合反应得到了具有抗菌性能的水凝胶。
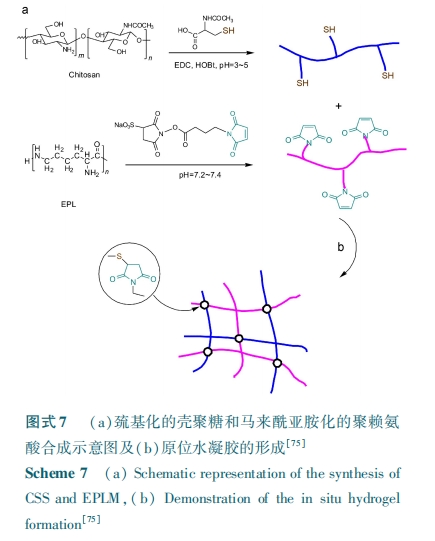
值得注意的是化学键交联的水凝胶很多采用了光交联技术,作为应用于生物医用领域的水凝胶材料,残留单体的去除以及引发剂或光敏剂的使用是需要给予关注的问题。目前大多数体系使用的光引发剂 Irgacure 2959 被认为是一种生物相容性较好的引发剂[55,70,76,77],体外细胞培养实验证明低剂量Irgacure 2959 引发交联的水凝胶对哺乳动物细胞并没有毒性[77]。同时,也有报道利用有机溶剂萃取、离心分离沉淀的方法来去除残留单体[38],以及对交联的凝胶进行浸泡冲洗的方法以尽可能去除残留的小分子物质[78]。
4 物理/ 化学双重交联水凝胶
相对于上述单一物理交联凝胶和化学交联凝胶,近年来出现了含多肽自组装的物理/ 化学双重交联水凝胶。在这类的体系中,多肽不但参与了凝胶网络的构建和对网络结构的控制,而且还赋予了水凝胶某些功能,如酶降解和促组织修复等。最重要的是多肽自组装贡献的物理交联结构还能对化学交联凝胶网络起到增强作用。多肽的引入确实给此类凝胶带来了诸多优良特性,然而这类水凝胶体系中需要注意:为充分发挥多肽的物理性能,引入的化学交联不能过度干扰体系中的多肽自组装。
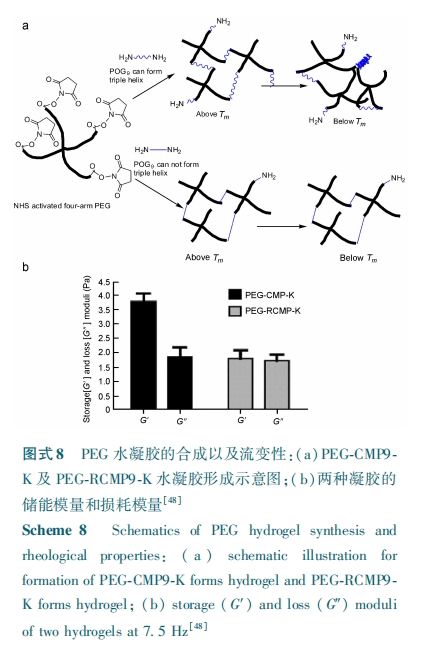
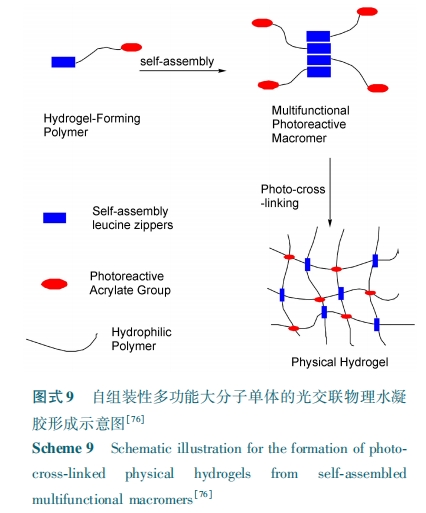
在这类物理/ 化学复合凝胶中,基于多肽自组装形成的物理交联结构不仅赋予了水凝胶特殊功能性,而且还可提高化学交联凝胶的力学性能。但由于化学交联网络的束缚不利于多肽的自组装,使得相关的工作开展得还很少,考虑到这类凝胶综合性能和功能的提升,未来预期这方面的工作会有更长远发展。
5 结论
多肽分子由于具有许多优良性能,比如生物相容性好、生物可降解、细胞黏附性、抗菌性以及自组装性,使得其成为近年来的研究热点。但另一方面,多肽可能的免疫原性也一直被给予高度关注,然而至今没有确切的答案。基于 Ryan 综述中提出的观点可以推断[81],如果体系中的多肽分子量较小或在相当短时间内就可被生物体清除,则其潜在的免疫原性较小甚至可以忽略,相反就需要考虑其对生物体的可能免疫原性。正如本综述所总结,基于多肽的优良性能可得到许多性能特殊的水凝胶,在医用领域有着广阔的应用前景。尽管含有多肽组分的聚合物水凝胶已有大量的研究,但我们认为这方面还有许多亟需解决的问题,比如基于多肽自组装的物理凝胶力学性能不好以及其对外界的刺激响应能力不强、具有潜在特殊功能的多肽亟待发现等等。虽面临诸多挑战,也恰恰为研究者提供了更多的研究空间,比如将化学交联引入基于多肽自组装的物理水凝胶中,但化学交联又不可过多干扰多肽自组装;多肽自组装形式和机理进行更深入研究对于根据特殊实际需求设计水凝胶结构具有重要指导意义。相信随着研究者的不懈努力,基于多肽的聚合物水凝胶将会在生物医用领域得到更长足的发展。
免责声明:本文为行业交流学习,版权归原作者及原杂志所有,如有侵权,可联系删除。文章标注有作者及文章出处,如需阅读原文及参考文献,可阅读原杂志。

电话:0551-65177703 邮箱:pb@peptidesbank.com 地址:安徽省合肥市四川路868号云谷创新园A6栋3层
合肥肽库生物(Taikubio)只为有资质的科研机构、医药企业基于科学研究或药证申报的用途提供医药研发服务, 不为任何个人或者非科研性质的、非用于药证申报使用等其他用途提供服务。