
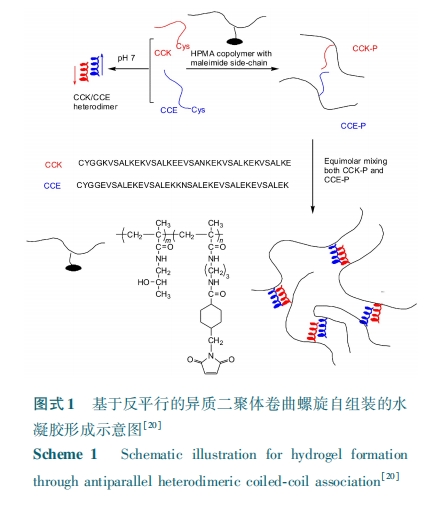
еЊ вЊ ЖрыФгЩгкОпгаСМКУЕФЩњЮяЯрШнадКЭЩњЮяПЩНЕНтадЁЂЩњЮяЛюадвдМАздзщзАЬиадЃЌ НќФъРДЪмЕНСЫЙуЗКЕФЙизЂЁЃНЋЖрыФздзщзАЬиадв§ШыЕНОлКЯЮяжаЃЌПЩИГгшОлКЯЮяаЮГЩФ§НКадВЂЖдФ§НКЭјТчЗжзгНсЙЙзіГівЛЖЈПижЦЃЌНјЖјЪЙФ§НКОпгаШчЛЗОГЯьгІЁЂСІбЇПЩЕїЕШНсЙЙПижЦадФмЃЛНЋЬиЪтЙІФмадЖрыФв§ШыЕНЛЏбЇНЛСЊЕФОлКЯЮяФ§НКЭјТчжаЃЌПЩИГгшЫЎФ§НКЩњЮяЙІФмадЃЌШчЯИАћ№ЄИНЁЂУИНЕНтЁЂПЙОњЕШЃЛНЋЖрыФЕФФ§НКЭјТчЙЙНЈЁЂНсЙЙПижЦзїгУвдМАЙІФмадЭЌЪБв§ШыЛёЕУЕФЮяРэЃЏ ЛЏбЇЫЋжиНЛСЊФ§НКВЛНіИГгшЫЎФ§НКвЛЖЈЕФЙІФмадЃЌЧвЖрыФздзщзАЙБЯзЕФЮяРэНЛСЊНсЙЙЛЙФмЖдЛЏбЇНЛСЊФ§НКЭјТчЦ№діЧПзїгУЁЃБОЮФзлЪіСЫЛљгкЖрыФздзщзАЕФЮяРэНЛСЊОлКЯЮяЫЎФ§НКЁЂЖрыФЙІФмЛЏЕФЛЏбЇНЛСЊОлКЯЮяЫЎФ§НКвдМАЛљгкЖрыФЕФЮяРэЃЏ ЛЏбЇЫЋжиНЛСЊЕФОлКЯЮяЫЎФ§НКЃЌВЂеЙЭћСЫетаЉЫЎФ§НКЕФЗЂеЙЧАОАЁЃ
ЫЎФ§НКЪЧгЩШ§ЮЌНЛСЊЭјТчНсЙЙКЭНщжЪЫЎЙВЭЌзщГЩЕФЖрдЊЬхЯЕЃЌЦфНЛСЊЭјТчжаКЌгаДѓСПЕФЧзЫЎадЛљЭХЃЌЕБЫЎЗжзгРЉЩЂЕНФ§НКЭјТчжаЪБЃЌЫЎФ§НКЮќЫЎКѓЬхЛ§ПЩХђеЭжСдРДЪ§БЖЁЃдкЙ§ШЅМИЪЎФъРяЃЌЫЎФ§НКгЩгкОпгаКЌЫЎСПИпЁЂНсЙЙгыЯИАћЭтЛљжЪЯрЫЦвдМАЩњЮяЯрШнадСМКУЕШЬиЕуЃЌЦфдквЉЮяЪЭЗХЃлЃБЃнЁЂзщжЏЙЄГЬЃлЃВЃнЁЂЯИАћХрбјЃлЃГЃнвдМАЩњЮя№ЄКЯЃлЃДЃЌЃЕЃн ЕШвНгУСьгђЕУЕНСЫЙуЗКгІгУЁЃ
здШЛНчДцдкЕФЬьШЛАБЛљЫсНіга ЃВЃА жжЃЌвЛаЉЗЧЬьШЛАБЛљЫсвВПЩЪЕбщКЯГЩЕУЕНЃЌЫќУЧОпгаВЛЭЌЕФЮяЛЏаджЪЃКМЋадЁЂЫсМюадвдМАЪЧЗёКЌЗМЯуЛљЭХЁЃгаЯожжРрЕФАБЛљЫсЭЈЙ§ыФМќАДвЛЖЈађСаХХСаПЩЕУЕНДѓСПОпгаВЛЭЌРэЛЏаджЪЕФЖрыФЃлЃЖЃнЁЃгЩгкЖрыФСДЖЮЩЯКЌгаДѓСПыФМќвдМАВЛЭЌНсЙЙЕФАБЛљЫсВаЛљЃЌЪЙЦфПЩРћгУБЫДЫМфЕФЧтМќЕШЗЧЙВМлМќзїгУРДЪЕЯжЗжзгЕФгааЇздзщзАЃЌБШШчОэЧњТна§НсЙЙЁЂІТ⁃елЕўНсЙЙЁЂШ§ЙЩТна§НсЙЙвдМАИќИпМЖНсЙЙЁЃНЋЖрыФздзщзАЬиадв§ШыЕНОлКЯЮяжаЃЌЪЙЕУдкФЩУзЫЎЦНЩЯгаађПижЦЦфЗжзгНсЙЙГЩЮЊПЩФмЃлЃЗЃЌЃИЃнЃЌЭЌЪБвВИГгшОлКЯЮяФ§НКвдгХСМЬиадЃЌБШШчЭтНчДЬМЄЯьгІадЁЂСІбЇПЩЕїадЁЃЖрыФЛЙгаЦфЫћЬиЪтЙІФмадЃКЯИАћ№ЄИНадЁЂЩњЮяПЩНЕНтадЁЂПЙОњадЕШЃЌБШШчКЌОЋАБЫс⁃ИЪАБЫс⁃ЬьЖЌАБЫсЃЈЃСЃђЃч⁃ЃЧЃьЃљ⁃ЃСЃѓЃ№ЃЌЃвЃЧЃФЃЉ ађСаЕФЖрыФФмИФЩЦЫЎФ§НКЕФЯИАћ№ЄИНадЃлЃЙЃнЁЃБОЮФИљОнЖрыФдкФ§НКЬхЯЕжаЕФВЛЭЌзїгУеыЖдНќЦкЛљгкЖрыФНсЙЙЕФИїРрЫЎФ§НКНјааСЫзлЪіЃЌВЂЖдбаОПЙ§ГЬжаГіЯжЕФЮЪЬтзїСЫМђвЊЗжЮіЁЃ
ЃВ ЮяРэНЛСЊЫЎФ§НК
ЫЎФ§НКЛљгкНЛСЊМќаджЪВЛЭЌЃЌЗжЮЊЛЏбЇЛђЮяРэФ§НКЁЃЛЏбЇЫЎФ§НКЪЧвЛжжгРОУадФ§НКЃЌ МДФ§НКЙ§ГЬВЛПЩФцЃЌЖјЮяРэФ§НКПЩЭЈЙ§ЗЧЙВМлМќЯрЛЅзїгУЕШЮяРэЗНЗЈНЛСЊЕУЕНЃЌЦфФ§НКЙ§ГЬОпгаПЩФцадЁЃЖрыФПЩРћгУОВЕчзїгУЁЂЪшЫЎзїгУЁЂІа⁃Іа ЖбЛ§вдМАЧтМќзїгУЕШздзщзАГЩВЛЭЌЕФЖўМЖЛђИпМЖНсЙЙЃЌБШШчОэЧњТна§ЃЈЃуЃяЃщЃьЃхЃф ЃуЃяЃщЃьЃЉЁЂ ІТ⁃елЕў ЃЈ ІТ⁃ЃѓЃшЃхЃхЃєЃЉЁЂ Ш§ ЙЩ Тн а§ ЃЈ ЃєЃђЃщЃ№ЃьЃхЃшЃхЃьЃщЃјЃЉЃЌЧвзщзАЙ§ГЬОпгаПЩФцадЃлЃИЃнЁЃНЋетаЉПЩЮяРэзщзАЕФНсЙЙв§ШыЕНОлКЯЮяЬхЯЕжаЃЌОЭЛсЪЙОлКЯЮяаЮГЩЮяРэФ§НКЭјТчНсЙЙЃЌЧвЖрыФзщзАЕФБОжЪЬиадЛЙЛсЭЌЪБИГгшЫЎФ§НКвдЛЗОГЯьгІЁЂСІбЇЕїПиЕШЖржжНсЙЙПижЦадФмЁЃ
ЃВ.ЃБ ЛљгкОэЧњТна§
ВЛЙмдкздШЛНчЛЙЪЧдкЪЕбщЪвжаЃЌОэЧњТна§ЕФГЌЖўМЖНсЙЙаЮГЩЖМЪЧЭЈЙ§СНИіЛђЖрИі ІС⁃Тна§ЖрыФЗжзгдкЫЎШмвКвдЬиЖЈЗНЪНРІАѓРДЭъГЩЕФЃЌетжжРІАѓЬиаджївЊ РД зд ІС⁃Тн а§ Жр ыФ Зж зг ЕФ Цп ыФ жи ИД ађ СаЃЈЃсЃтЃуЃфЃхЃцЃчЃЉЃю НсЙЙЃЌЦфЖрыФЗжзгжиИДађСажаЕФ ЃсЁЂЃфЁЂЃхЁЂЃч ЮЛжУЕФАБЛљЫсВаЛљжЎМфЕФЯрЛЅзїгУЪЧЙЙНЈОэЧњТна§ЕФФкдкзїгУСІЃЌЦфжаЮЛжУ Ѓс КЭ Ѓф жївЊгЩЪшЫЎЕФАБЛљЫсВаЛљЫљеМОнЃЌЗжзгМфжївЊЮЊЪшЫЎадзїгУСІЃЌЖјОэЧњТна§НсЙЙФкВПЕФЪшЫЎадЧПШѕжївЊгЩ Ѓх КЭ Ѓч ЮЛжУЕФАБЛљЫсВаЛљжЎМфЕФОВЕчзїгУРДЕїНкЃЌЖд ІС⁃Тна§ЕФЮШЖЈадОпгаживЊЕФзїгУЁЃ
дчдк ЃБЃЙЃЙЃИ ФъЃЌЃаЃхЃєЃыЃс ЕШЃлЃБЃАЃн ОЭРћгУ ЃФЃЮЃС жизщММЪѕЩшМЦСЫвЛжжШ§ЧЖЖЮЙВОлЮяЃКЙВОлЮяЖЫЭЗЮЊССАБЫсРСДНсЙЙЃЌжаМфЮЊЫЎШмадЁЂЗжзгСДЮоЙцОэЧњЕФОлЕчНтжЪЁЃЦфжаССАБЫсРСДНсЙЙгђПЩаЮГЩОэЧњТна§ОлМЏгђЖјДйЪЙОлКЯЮяШ§ЮЌЭјТчНсЙЙаЮГЩЃЌЕУЕНСЫОпгаЭтНчЯьгІадЕФЫЎФ§НКЁЃРћгУИУЩшМЦЗНЗЈЃЌЃиЃѕ ЕШЃлЃБЃБЃн вВЩшМЦСЫвЛЯЕСаЕФШ§ЧЖЖЮЙВОлЮяЃЌжЛЬцЛЛСЫЖЫЭЗЕФССАБЫсРСДНсЙЙЃЌжаМфСДЖЮШдЮЊЧзЫЎЕФОлЕчНтжЪЧјгђЃЌЕУЕНЕФЫЎФ§НКЭЌбљФмЙЛЯьгІЭтНчЛЗОГЕФДЬМЄЃЌЧвСНЖЫЭЗОэЧњТна§гђЕФАБЛљЫсађСаЖдЫЎФ§НКЕФНсЙЙКЭаджЪгазХживЊгАЯьЁЃЛЙгабаОПепЭЈЙ§ПижЦДЫРрЗжзгЕФЭјТчЭиЦЫНсЙЙРДДяЕНЕїНкИУЫЎФ§НКЧжЪДТЪЕФФПЕФЃЌжївЊдкгкбЙжЦСЫЛЗзДЗжзгаЮГЩЃлЃБЃВЃнЁЃвдЩЯЕФЬхЯЕЖМВЛКЌШЫЙЄКЯГЩОлКЯЮяЃЌЮЊСЫНЋ ІС⁃Тна§ЖрыФКЭОлКЯЮяСНепгХСМадФмНсКЯдквЛЦ№ЃЌгабаОПепНЋОэЧњНсЙЙв§ШыЕНОлввЖўДМЃЈЃаЃХЃЧЃЉЗжзгжаЃЌЗЂЯжОэЧњТна§ЕФздзщзАФмСІвРШЛДцдкЃЌЧвЭЈЙ§ПижЦАБЛљЫсЕФађСаПЩПижЦДЫРрЙВОлЮяЕФНсЙЙКЭадФмЃлЃБЃГ ЁЋ ЃБЃЕЃнЁЃвдЩЯЕФОэЧњТна§жївЊРДздЗЧШЫРрЯИАћФкЕФЖрыФЃЌЮЊСЫМѕЩйДЫРрВФСЯгУгкЬхФкЪБЫљв§Ц№ЕФУтвпдадЃЌЃЪЃщЃюЃч ЕШЃлЃБЃЖЃн ЖдРДдДгкШЫЯЫЮЌЕААзТна§ЧјЕФЖрыФађСажаЬиЖЈЮЛжУАБЛљЫсВаЛљзїСЫЬцЛЛЃЌВЂНЋЦфНгжІЕНСЫ ЃаЃХЃЧ ЗжзгЕФСНЖЫЃЌЕУЕНЕФШ§ЧЖЖЮЙВОлЮядкЖрыФаЮГЩЖўОлЬхЛђЫФОлЬхзїгУЯТЕУЕНСЫ№ЄЕЏадЕФЮяРэФ§НКЁЃ
Г§СЫЧЖЖЮЙВОлЮяЃЌЛљгкОэЧњТна§здзщзАЕФНгжІЙВОлЭЌбљвВПЩЕУЕНадФмВЛЭЌЕФЮяРэЫЎФ§НКЁЃЦфжаЃЫЃяЃ№ЃхЃуЁІ ЃхЃы ПЮЬтзщзіСЫБШНЯЯЕЭГЕФбаОПЃКНЋПЩаЮГЩОэЧњТна§ЕФЖрыФНгжІЕНЛљгк ЃЮ⁃ЃЈЃВ⁃єЧБћЛљЃЉМзЛљБћЯЉѕЃАЗЃЈЃШЃаЃЭЃСЃЉЙВОлЮяЕФжїСДЩЯЃЌЗЂЯжЖрыФГЄЖШКЭЪ§СПЖдЙВОлЮяГЩНКгаживЊгАЯьЃлЃБЃЗЃнЃЌНјвЛВНбаОПЗЂЯжгАЯьЙВОлЮяздзщзАЖЏСІбЇЕФжївЊвђЫиЪЧХЈЖШКЭЮТЖШЃлЃБЃИЃнЁЃдчдкЪЎМИФъЧАЃЌИУПЮЬтзщОЭБЈЕРСЫЭЈЙ§Н№ЪєТчКЯзїгУНЋПЩаЮГЩОэЧњТна§ЕФЕААзжЪЛљађНгжІЕНЛљгк ЃШЃаЃЭЃС ЙВОлЮяжїСДЩЯЃЌЕУЕНСЫФ§НКЭпНтЮТЖШКЭОэЧњТна§ЕФНтСДЮТЖШвЛжТЕФЫЎФ§НКЃЌЧвФ§НКЮТУєадЛЙПЩЭЈЙ§ЬижЦЛљвђЙЄГЬЕААзКЭЖрыФЕФДцдкРДПижЦЃлЃБЃЙЃнЁЃдкетжжЬхЯЕжаЕФЮяРэНЛСЊНсЙЙЪЧИДдгЕФЃЌЦфжаЭЌдДЖўОлЬхЪЧвЛжжЮоаЇНЛСЊНсЙЙЃЌЪЙЕУЬхЯЕЕФНЛСЊУмЖШЪмЕНКмДѓгАЯьЁЃЮЊСЫНтОіетвЛЮЪЬтЃЌИУПЮЬтзщгжЭЈЙ§ТѕПЫЖћМгГЩЗДгІНЋДјгаЯрЗДЕчКЩЕФКЯГЩЖрыФЃЈЃУЃУЃХ КЭ ЃУЃУЃЫЃЉЗжБ№МоНгЕНСЫЛљгкОл ЃЮ⁃ЃЈЃВ⁃єЧБћЛљЃЉМзЛљБћЯЉѕЃАЗЃЈЃаЃШЃаЃЭЃСЃЉЕФЙВОлЮяЕФВрСДЩЯЕУЕНСЫЃУЃУЃХ⁃Ѓа КЭ ЃУЃУЃЫ⁃ЃаЃЈЃа ЮЊ ЃШЃаЃЭЃС ЕФжїСДЃЉЁЃдкжаад Ѓ№ЃШЬѕМўЯТЃЌНЋЕШФІЖћ ЃУЃУЃЫ⁃Ѓа КЭ ЃУЃУЃХ⁃Ѓа ЛьКЯЃЌгЩгкДјЯрЗДЕчКЩЕФ ЃУЃУЃХ КЭ ЃУЃУЃЫ МфЕФОВЕчЮќв§аЮГЩЕФЗДЦНаавьжЪЖўОлЬхЦ№ЕНСЫЮяРэНЛСЊзїгУЃЌЖј ЃУЃУЃЫ Лђ ЃУЃУЃХМфгЩгкОВЕчЕФХХГтЖјВЛФмаЮГЩЭЌдДЖўОлЬхЃЌЪЙЦфдкКмЕЭХЈЖШЯТОЭПЩгааЇаЮГЩЫЎФ§НКЃЌШчЭМЪН ЃБЁЃЖјЕЅЖР ЃУЃУЃЫ⁃Ѓа Лђ ЃУЃУЃХ⁃Ѓа НгжІЙВОлЮяШДВЛФмаЮГЩЫЎФ§НКЁЃЕБЯђетжжЫЎФ§НКЬхЯЕжаМгШывЛЖЈСПЕФБфадМСбЮЫсывЪБЃЌгЩгкОэЧњТна§НсЙЙгђЕФБфадЃЌЦЦЛЕСЫЮяРэНЛСЊНсЙЙЪЙЕУЫЎФ§НКШмНтЃЌЖјЕБАббЮЫсывГ§ШЅКѓЫЎФ§НКжиаТаЮГЩЁЃетжжЫЎФ§НКЕФаджЪКЭЭјТчНсЙЙЛЙПЩЭЈЙ§ИФБфНгжІЙВОлЮяжїСДЕФОлКЯЮяКЭОэЧњТна§ЖрыФЕФНсЙЙМАЪ§СПРДЕїНкЃлЃВЃАЃнЁЃМјгк ІС⁃ОэЧњТна§ЕФЖРЬиЮяРэНЛСЊНсЙЙвдМАЖрыФБОЩэЕФгХСМЬиадЃЌетРрЫЎФ§НКПЩзїЮЊвЉЮядиЬхЕФРэЯыКђбЁВФСЯЃлЃВЃБ ЁЋ ЃВЃЕЃнЁЃ
ЃВ. ЃВ Лљгк ІТ⁃елЕў
ВЛЭЌгкАєзДЕФ ІС⁃Тна§ЖўМЖНсЙЙЃЌІТ⁃елЕўЪЧгЩЖрыФСДЭЈЙ§ ЃЮЃШ КЭ ЃУ=ЃЯ жЎМфЕФЧтМќзїгУСІаЮГЩЕФЦНааЛђЗДЦНааЗНЪНХХСаЕФБЁЦЌЃЌЦфФкВПзїгУСІжївЊЪЧЖрыФСДЖЮМфЩЯыФМќМфЕФЧтМќзїгУЁЃІТ⁃елЕўздзщзАЪЧЭЈЙ§ЗжзгМфвдМАЗжзгФкЕФзїгУСІЧ§ЪЙЕФЃЌБШШчОВЕчзїгУКЭЪшЫЎзїгУЃЌвђДЫЦфздзщзАЕФЙ§ГЬФмЙЛЯьгІЭтНчЕФБфЛЏЁЃ
Й§ШЅаэЖрЛљгк ІТ⁃елЕўздзщзАЕФЧЖЖЮЙВОлЮявбБЛЙуЗКбаОПЁЃНЋ ІТ⁃елЕўЖрыФНгжІЕНЧзЫЎЕФОлввЖўДМЃЈЃаЃХЃЧЃЉЃлЃВЃЖ ЁЋ ЃГЃБЃнвдМА ЃаЃШЃаЃЭЃС ЩЯЃлЃГЃВЃнЃЌЗЂЯжЖрыФШдБЃСєзХЦфЙЬгаЕФздзщзАФмСІЃЌЛЙПЩздзщзАГЩИїжжФЩУзвдМАЮЂУзНсЙЙЃЌЕЋОлКЯЮяДцдквВМѕШѕСЫЖрыФЖдЭтНчДЬМЄЕФУєИаадЁЃдкетаЉбаОПЛљДЁЩЯЃЌ ЃЫЃяЃ№ЃхЃуЁІ ЃхЃы ПЮЬтзщЃлЃГЃГЃн ЭЈ Й§ ЛЏ бЇ ао ЪЮ НЋ ПЩ аЮ ГЩ ІТ⁃ел Еў ЕФ Жр ыФЃдЃдЃвЃЦЃдЃзЃдЃЦЃдЃдЃдЃЈЃТЃхЃєЃсЃБЃБЃСЃЉ НгжІЕНСЫЛљгк ЃШЃаЃЭЃС ЙВОлЮяЕФжїСДЩЯЃЌЦфжаВрСДЕФ ЃТЃхЃєЃсЃБЃБЃС аЮГЩЕФЖрМЖ ІТ⁃елЕўНсЙЙЦ№ЕНСЫЮяРэНЛСЊЕФзїгУЁЃЕУЕНЙВОлЮяЬхЯЕЕФГЩНКЪБМфОпгаУїЯдЕФХЈЖШвРРЕадЃКОлКЯЮяЕФХЈЖШЮЊ ЃГ ЃїЃєЃЅ ЪБЃЌЦфдкЪ§ЗжжгФкМДПЩГЩНКЃЌЖјдкХЈЖШНЯЕЭЪБЃЌГЃЮТЯТашвЊЪ§ЬьВХПЩГЩНКЁЃГ§ДЫжЎЭтЃЌКЯГЩОлКЯЮяЕФЦСБЮаЇгІЪЙЕУЖрыФЖдЮТЖШКЭ Ѓ№ЃШ ЕФБфЛЏУєИаадНЕЕЭЁЃИУЬхЯЕжЛФмдкЫсадЕФЬѕМўЯТаЮГЩЯЫЮЌНсЙЙЃЌЮЊСЫЪЙЦфдкжаадЬѕМўЯТОЭПЩаЮГЩЫЎФ§НКЃЌИУПЮЬтзщНЋЛЅ ВЙ ЕФ Жр ыФ ЃдЃдЃвЃЦЃдЃзЃдЃЦЃдЃдЃд КЭ ЃдЃдЃХЃЦЃдЃзЃдЃЦЃХЃдЃдЃЈЃТЃхЃєЃсЃБЃБЃС КЭ ЃТЃхЃєЃсЃБЃБЃТЃЉНгжІЕНСЫЛљгк ЃШЃаЃЭЃС ЙВОлЮяЕФжїСДЩЯЃлЃГЃДЃнЃЌЭЌЪБЮЊСЫЬсИпВФСЯЕФЯИАћ№ЄИНадЃЌНЋЃвЃЧЃФ вВНгжІЕНСЫЙВОлЮяЕФВрСДЩЯЃЌДгЖјЕУЕНСЫ Ѓ№ЃяЃьЃљЃЈ ЃШЃаЃЭЃС ЃЉ⁃Ѓч⁃ЃТЃхЃєЃсЃБЃБЃС⁃ЃвЃЧЃФ КЭ Ѓ№ЃяЃьЃљ ЃЈ ЃШЃаЃЭЃС ЃЉ⁃Ѓч⁃ЃТЃхЃєЃсЃБЃБЃТ⁃ЃвЃЧЃФЁЃНЋ СН жж ЃвЃЧЃФ Нг жІ Юя АД ЃТЃхЃєЃсЃБЃБЃСЃКЃТЃхЃєЃсЃБЃБЃТ ЃН ЃБ ЁУ ЃБ ЕФ БШ Р§ Ль КЯЃЌ жа ад Ьѕ Мў ЯТ ЛЅ ВЙ ЕФЃТЃхЃєЃсЃБЃБЃС КЭ ЃТЃхЃєЃсЃБЃБЃТ аЮГЩЕФЖрМЖЗДЦНааЕФ ІТ⁃елЕўНсЙЙЦ№ЕНСЫЮяРэНЛСЊЕФзїгУЃЌШчЭМЪН ЃВЁЃбаОПЗЂЯжОлКЯЮяЕФХЈЖШЮЊ ЃБ ЃїЃєЃЅ ЁЋ ЃГ ЃїЃєЃЅ ЪБЃЌЦфГЩНКЪБМфдк ЃВ ЁЋЃВЃД Ѓш ФкБфЛЏЃЌЧветжжНгжІЙВОлЮяЬхЯЕжааЮГЩЕФФЩУзЯЫЮЌНсЙЙдкФЃФтЬхвКжаФмЙЛаЮГЩРрєЧСзЛвЪЏЕФОЇЬхЃлЃГЃЕЃнЃЌЫфШЛетжжЭъШЋЛљгкЮяРэНЛСЊЕФЫЎФ§НКСІбЇадФмВЛКУЃЈХЈЖШЮЊЃГ ЃїЃєЃЅ ЃЌЦфДЂФмФЃСПдМЮЊЃДЃАЃА ЃаЃсЃЉЃЌЕЋЦфФкВПИїЯђвьадЕФЖрПзНсЙЙЃЌЪЙЦфдкЙЧдйЩњЙЄГЬжЇМмгІгУЗНУцгазХЧБдкЕФМлжЕЁЃ
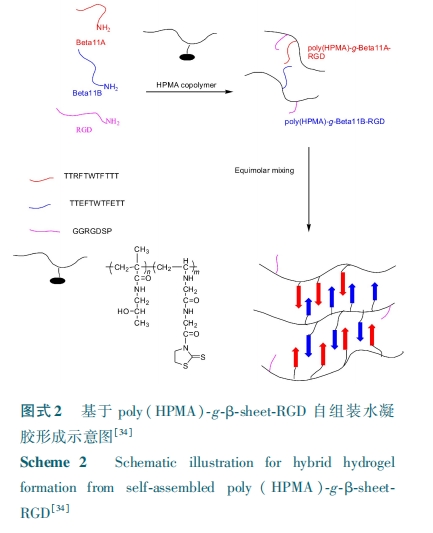
ЭИУїжЪЫсгазХЙуЗКЕФгІгУЃЌПЩвдЕїНкЦфСїБфбЇаджЪРДТњзуИїжжгІгУЃЌЃХЃьЃфЃхЃђ ЕШЃлЃГЃЖЃн НЋ ЃЬЃгЃЬЃгЃЬЃгЃЬЃг ЖрыФНгжІдкЭИУїжЪЫсЃЈЃШЃСЃЉЕФжїСДЩЯЃЌаЮГЩЕФЖрМЖ ІТ⁃елЕўНсЙЙдк ЃШЃС ЗжзгСДМфЦ№ЕНСЫЮяРэНЛСЊзїгУЃЌКЭУЛгаЖрыФаоЪЮЕФ ЃШЃС ЯрБШЃЌІТ⁃елЕўУїЯдИФЩЦСЫ ЃШЃС ЕФЕЭМєЧа№ЄЖШЁЃ
вВгабаОПепНЋ ІТ⁃елЕўНсЙЙв§ШыЕНОпгаЮТУєадЕФОл ЃЮ⁃вь Бћ Лљ Бћ ЯЉ ѕЃ АЗ ЃЈ ЃаЃЮЃЩЃаЃСЃСЃэ ЃЉ ЕФ Ьх ЯЕжаЃлЃГЃЗ ЁЋ ЃГЃЙЃнЃЌЃЭЃщЃьЃьЃхЃђ ПЮ Ьт зщ зю ПЊ ЪМ НЋ ЖЫ ЭЗ лЯ Лљ ЛЏ ЕФЃЦЃХЃЦЃХЃЦЃЫЃЦЃЫ АЫыФзїЮЊздгЩЛљОлКЯЕФ ЃаЃЮЃЩЃаЃСЃСЃэ ЕФСДзЊвЦМСЃЌНЋетжжАЫыФНгжІЕНСЫ ЃаЃЮЃЩЃаЃСЃСЃэ ЩЯЃЌЕУЕНСЫОпгаЫЋЮТЖШЯьгІадЕФЫЎФ§НКЃЌетЮЊКЯГЩЖрыФКЭЮТУєадОлКЯЮяЕФаТаЭЮТУєадДѓЗжзгПЊБйСЫаТЕФЭООЖЁЃДПЃаЃЮЃЩЃаЃСЃСЃэ ВЛФмаЮГЩЮШЖЈЕФЮяРэФ§НКЃЌЮЊСЫБмУтЛЏбЇНЛСЊМСЖдзщжЏЕФЖОИБзїгУЃЌИУПЮЬтзщЭЈЙ§СДзЊвЦЗДгІ НЋ ВЛ ЭЌ КЌ СП ЕФ ЃаЃЮЃЩЃаЃСЃСЃэ⁃ЃЦЃХЃЦЃХЃЦЃЫЃЦЃЫ в§ Шы ЕНЃаЃЮЃЩЃаЃСЃСЃэ жаЃЌЕїНкСНепБШР§РДИФБфЦфЬхЯЕжаЕФздзщзАГЬЖШЃЌНјЖјПижЦФ§НКЭјТчжаЕФЮяРэНЛСЊУмЖШЃЌДгЖјЕУЕНСЫСІбЇадФмПЩЕїЕФЫЎФ§НКЁЃДЫЭтЃЌЮЊСЫИГгшУЛгазюЕЭСйНчШмвКЮТЖШЕФ ЃЦЃХЃЦЃХЃЦЃЫЃЦЃЫ ЫЎФ§НКвдЮТУєадЃЌ ИУ ПЮ Ьт зщ гж НЋ ЃаЃЮЃЩЃаЃСЃСЃэ⁃ЃЦЃХЃЦЃХЃЦЃЫЃЦЃЫ Мг Шы ЕНЃЦЃХЃЦЃХЃЦЃЫЃЦЃЫ ЕФ Ьх ЯЕ жаЃЌ ба ОП ЗЂ Яж ЃаЃЮЃЩЃаЃСЃСЃэ Ыц зХЃаЃЮЃЩЃаЃСЃСЃэ⁃ЃЦЃХЃЦЃХЃЦЃЫЃЦЃЫ КЭ ЃЦЃХЃЦЃХЃЦЃЫЃЦЃЫ здзщзАБЛЙЬЖЈдкФЩУзЯЫЮЌНсЙЙЩЯЃЌЕЋетВЂУЛгагАЯь ЃаЃЮЃЩЃаЃСЃСЃэ ЕФзюЕЭСйНчШмвКЮТЖШЃЌЧв ЃаЃЮЃЩЃаЃСЃСЃэ⁃ЃЦЃХЃЦЃХЃЦЃЫЃЦЃЫ ЕФМгШыУїЯддіМгСЫЫЎФ§НКЕФМєЧаФЃСПЁЃЯрБШгкЕЅвЛЮТУєадФ§НКЃЌетРрСІбЇадФмПЩЕїЕФЮТУєадФ§НКгазХИќЙуРЋЕФгІгУЧАОАЁЃ
ЃВ.ЃГ ЛљгкШ§ЙЩТна§
НКдФЃФтЖрыФЃЈ ЃуЃяЃьЃьЃсЃчЃхЃю ЃэЃщЃэЃхЃєЃщЃу Ѓ№ЃхЃ№ЃєЃщЃфЃхЃЌЃУЃЭЃаЃЉЭЈГЃгЩ ЃБЃЕ ЁЋ ЃДЃЕ ИіАБЛљЫсВаЛљЙЙГЩЃЌЦфжажиИДШ§ыФЃЈЃЧЃьЃљ⁃ЃиЃсЃс⁃ЃйЃсЃсЃЉЃю ЕФДцдкЪЙЦфОпгаздЗЂаЮГЩШ§ЙЩТна§НсЙЙЕФЬиадЃЌЦфжа ЃиЃсЃс ЭЈГЃЮЊИЌАБЫсЃЈ ЃаЃђЃяЃЉЃЌЃйЃсЃс ЭЈГЃЮЊєЧИЌАБЫсЃЈЃШЃљЃ№ЃЉЁЃВЛЭЌгкЬьШЛЕФНКдЃЌШ§ЙЩТна§НтСДааЮЊОпгаЮТЖШПЩФцадЃЌЧвЦфНтСДЮТЖШПЩЭЈЙ§ЖрыФЕФСДГЄКЭАБЛљЫсзщГЩРДИФБфЁЃЙ§ШЅЪЎМИФъЃЌЫцзХЙЬЯрЖрыФКЯГЩММЪѕЕФЗЂеЙЃЌдкЪЕбщЪвОЭПЩРћгУГЩЪьЕФММЪѕКЯГЩОпгаЬиЖЈГЄЖШКЭЬиЖЈађСаНКдФЃФтЖрыФЁЃвЛаЉЕфаЭНКдФЃФтЖрыФЃЌБШШчЃЈЃЧЃьЃљ⁃ЃаЃђЃя⁃ЃШЃљЃ№ЃЉЃБЃАЃЈЃаЃЯЃЧЃБЃА ЃЉ КЭ ЃЈ ЃЧЃьЃљ⁃ЃаЃђЃя⁃ЃаЃђЃяЃЉЃБЃА ЃЈ ЃаЃаЃЧЃБЃА ЃЉ вВ вб ЩЬ вЕ ЛЏЁЃЃУЃЭЃа ЕФШ§ЙЩТна§НсЙЙЛЙПЩздзщзАГЩИќИпМЖЕФНсЙЙЃЌЃТЃђЃяЃфЃѓЃыЃљ ПЮЬтзщдјОБЈЕРЙ§НКдФЃФтЖрыФ ЃаЃЯЃЧЃБЃА ПЩзщзАГЩжІзДЕФЯИЫПНсЙЙЃлЃДЃАЃнЁЃ
ЕЅЖРЕФ ЃУЃЭЃа КмФбаЮГЩШШЮШЖЈЕФШ§ЙЩТна§НсЙЙЁЃЮЊСЫНтОіетвЛЮЪЬтЃЌИїжжЙВМлМќНсБЛгУгкЮШЖЈ ЃУЃЭЃаШ§ЙЩТна§НсЙЙЃлЃДЃБЃнЃЌБШШчАыызАБЫсНсЁЂШ§ЙйФмЖШгаЛњЮяЁЃЪїжІзДИпЗжзгЃЈ ЃфЃхЃюЃфЃђЃщЃэЃхЃђЃЉ ЭтЮЇБэУцКЌгаИпУмЖШЛљЭХЃЌЪЙЦфвВПЩзїЮЊЮШЖЈ ЃУЃЭЃа Ш§ЙЩТна§НсЙЙЕФЙВМлМќНсЁЃЃЫЃщЃюЃтЃхЃђЃчЃхЃђ ЕШЃлЃДЃВЃЌЃДЃГЃн НЋЪїжІзДЗжзгзїЮЊЮШЖЈШ§ЙЩТна§НсЙЙЕФЙВМлМќНсЃЌбаОПЗЂЯжЃЈЃЧЃьЃљ⁃ЃаЃђЃя⁃ЃЮЃьЃхЃѕЃЈЃЮ⁃ЃщЃѓЃяЃтЃѕЃєЃљЃьЃчЃьЃљЃуЃщЃюЃхЃЉЃЉаоЪЮЕФЪїжІзДЗжзгжааЮГЩСЫРрНКдЕААзЕФШ§ЙЩТна§НсЙЙЃЌетЮЊНјвЛВНРћгУ ЃУЃЭЃа здзщзАадРДаЮГЩЪїжІзДЫЎФ§НКЬсЙЉСЫбаОПЛљДЁЁЃЛљгкШ§ЙЩТна§НсЙЙЕФЮТЖШПЩФцадвдМАЦфздзщзАадЕУЕНЕФЫЎФ§НКЃЌЦфжаЖрыФЕФГЄЖШКЭађСазщГЩЖдзюКѓДѓЗжзгЕФНсЙЙгазХживЊЕФгАЯьЁЃЃЫЃяЃъЃщЃэЃс ЕШЃлЃДЃДЃн НЋЃЈЃЧЃьЃљ⁃ЃаЃђЃя⁃ЃаЃђЃяЃЉЃЕ ЃЈ ЃаЃаЃЧЃЕ ЃЉ НгЕНЫФДњОлѕЃАЗ⁃АЗаЭЪїжІзДИпЗжзгЃЈЃаЃСЃЭЃСЃЭЃЉЭтЮЇБэУцЕУЕНСЫ ЃаЃаЃЧЃЕ⁃ЃфЃхЃюЃЌгЩгкКмДѓЪ§СПЕФШ§ЙЩТна§НсЙЙДІгкЕЭЮШЖЈзДЬЌЃЌЪЙЕУЬхЯЕВЛФмаЮГЩзуЙЛЕФЮяРэНЛСЊЭјТчНсЙЙЃЌЫќжЛФмдкввДМКЭСђЫсФЦЕФДцдкЯТВХПЩаЮГЩЫЎФ§НКЁЃыФСДГЄЖШЖдШ§ЙЩТна§ЕФЮШЖЈадгазХживЊЕФгАЯьЃЌвђДЫПЩдіМгыФСДГЄЖШРДЬсИпШ§ЙЩТна§НсЙЙЮШЖЈадЃЌДгЖјдкУЛгаШЮКЮЬэМгМСЕФЧщПіЯТОЭПЩЪЙЦфзщзАГЩЫЎФ§НКЁЃЃгЃѕЃхЃшЃщЃђЃя ЕШЃлЃДЃЕЃнНЋЃЈЃЧЃьЃљ⁃ЃаЃђЃя⁃ЃаЃђЃяЃЉЃБЃАНгЕНЫФДњ ЃаЃСЃЭЃСЃЭ ЕФБэУцЃЌЕУЕНЕФЃаЃаЃЧЃБЃА⁃ЃфЃхЃю дкУЛгаШЮКЮЪдМСзїгУЯТОЭПЩаЮГЩЫЎФ§НКЃЌетЕУвцгк ЃаЃаЃЧЃБЃАдк ЃаЃСЃЭЃСЃЭ БэУцЕФЭХДиаЇгІвдМАыФСДГЄЖШЕФдіМгЃЌЕЋЦфФ§НКааЮЊРрЫЦгкУїНКЁЃ
ЩЯЪіЪЙгУЕФ ЃУЃЭЃа ЖМШБЗІєЧИЌАБЫсЃЌЪЙЕУ ЃУЃЭЃаШ§ЙЩТна§ЕФаЮГЩаЇТЪвдМАЮШЖЈадЖМКмЕЭЃЌЖјВЛФмгааЇДйНјШ§ЙЩТна§НсЙЙзщзАГЩИќИпМЖНсЙЙЁЃЃЫЃяЃъЃщЃэЃсЕШЃлЃДЃЖЃнНЋКЌгаєЧИЌАБЫсЃЈ ЃШЃљЃ№ЃЉ ЕФЃЈЃЧЃьЃљ⁃ЃаЃђЃя⁃ЃШЃљЃ№ЃЉЃБЃА НгжІЕНЫФДњ ЃаЃСЃЭЃСЃЭ БэУцЕУЕНСЫ ЃаЃЯЃЧЃБЃА⁃ЃфЃхЃюЃЌШчЭМЪН ЃГЃсЫљЪОЁЃВЛЭЌгк ЃаЃаЃЧЃБЃА⁃ЃфЃхЃюЃЌЦфФ§НКааЮЊРрЫЦгкЬьШЛНКдЖјЗЧУїНКЃЌМДЕБЫќМгШШжСЮТЖШдМ ЃДЃА Ёц ЪБЗЂЩњШмвК⁃Ф§НКзЊБфЃЌдкЕЭгк ЃВЃЕ Ёц ЪБЗЂЩњФ§НК⁃ШмвКзЊБфЁЃетПЩФмЪЧгЩгк ЃаЃСЃЭЃСЃЭ БэУцЕФШ§ЙЩТна§НсЙЙМДЪЙдкНЯЕЭЕФЮТЖШЯТвВФмздзщзАаЮГЩФЩУзСЃзгНсЙЙЃЌЧвгЩгкЧтМќзїгУЪЙЕУТна§НсЙЙжаКЌгаНЯЖрЕФЫЎЗжзгЁЃЕЋЕБЮТЖШЩ§ИпЪБЃЌШ§ЙЩТна§НсЙЙБфЕУЫЩЩЂЪЙЫЎЗжзгБЛДђТвВЂжиХХЃЌДгЖјгеЕМШ§ЙЩТна§здзщзАжТЪЙЬхЯЕШ§ЮЌЭјТчЕФаЮГЩЃЌШчЭМЪН ЃГЃт ЫљЪОЁЃЃаЃЯЃЧЃБЃА⁃ЃфЃхЃю ЫЎФ§НКЕФЕЏадФЃСПОпгаУїЯдЕФХЈЖШвРРЕадЃЌЕБ ЃаЃЯЃЧЃБЃА⁃ЃфЃхЃюЕФХЈЖШЗжБ№ЮЊЃЕ ЃїЃєЃЅ ЁЂЃБЃА ЃїЃєЃЅ ЁЂЃБЃЕ ЃїЃєЃЅЪБЃЌЦфФ§НКЖдгІЕФЕЏадФЃСПЗжБ№ЮЊ ЃВ ЃД ЁС ЃБЃАЃГЃаЃсЃЈЃЖЃА ЁцЃЉЁЂЃГ ЃБ ЁС ЃБЃАЃГЃаЃсЃЈЃЖЃГ Ёц ЃЉКЭ ЃБ ЃГ ЁС ЃБЃАЃДЃаЃс ЃЈЃЕЃА Ёц ЃЉЁЃгЩгкетРрЫЎФ§НКОпгаЬиЪтЕФЮТУєадЃЌЪЙЦфПЩзїЮЊвЛжжжЧФмвЉЮяЪЭЗХдиЬхЁЃЃЫЃяЃъЃщЃэЃс ЕШЃлЃДЃЗЃн гжЬжТлСЫШчКЮИФЩЦДЫРрЫЎФ§НКЮТЖШЯьгІадЕФвЉЮяЪЭЗХЃЌЪЕбщжаНЋФЃФтЖрыФ ЃаЃЯЃЧЃюЃЈЃю ЃН ЃВЃЌЃЕЃЌЃИЃЌЃБЃАЃЉНгЕНЫФДњ ЃаЃСЃЭЃСЃЭ ЕФБэУцЩЯЕУЕНСЫЃаЃЯЃЧЃю⁃ЃфЃхЃюЃѓЃЌбаОПВтЪдБэУїЃЌ ЃаЃЯЃЧЃю ИќФмдк ЃаЃСЃЭЃСЃЭ ЕФБэУцгааЇЕигеЕМШ§ЙЩТна§НсЙЙЕФаЮГЩЃЌЧвыФСДГЄЖШНЯГЄЕФ ЃаЃЯЃЧЃю ОпгаИќИпЕФШ§ЙЩТна§аЮГЩаЇТЪвдМАИќКУЕФШШЮШЖЈадЁЃЪЕбщжавдУЕЙхКьзїЮЊФЃаЭвЉЮяЃЌбаОПЗЂЯж ЃаЃЯЃЧЃю⁃ЃфЃхЃюЃѓ ИФЩЦСЫФЃаЭвЉЮядк ЃГЃЗ Ёц ЕФБЃСєТЪЃЌЯр БШ гк ЃаЃаЃЧЃю Жр ыФЃЌ ЪЙ Цф Иќ ЪЪ КЯ вЉЮяЕФЪЭЗХгІгУЁЃ
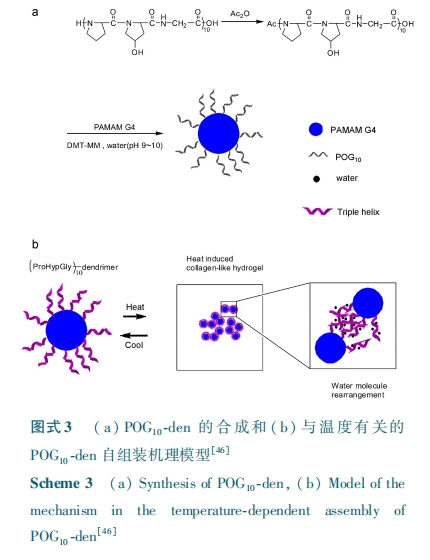
Г§СЫгІгУгквЉЮяЪЭЗХЭтЃЌЛљгк ЃУЃЭЃа здзщзАЕФИДКЯЫЎФ§НКЛЙПЩзїЮЊжЇМмВФСЯЁЃЦфжазщжЏжЇМмЕФСІбЇадФмЖдЯИАћЕФаЮЬЌКЭЗжЛЏгазХживЊЕФгАЯьЃЌЯЃЭћЦфСІбЇадФмПЩИљОнОпЬхЕФашвЊФмгааЇЕФЕїНкЁЃЮЊСЫНтОіетвЛЮЪЬтЃЌЃйЃѕ ПЮЬтзщЃлЃДЃИЃн ШЯЮЊЭЈЙ§ПижЦШ§ЙЩТна§ЕФЩњГЩгыЦЦЛЕПЩДяЕНЕїПиЫЎФ§НКСІбЇадФмЕФФПЕФЁЃЪЕбщжаНЋФЃФтЖрыФ ЃаЃЯЃЧЃЙ ЛЏбЇаоЪЮЕНСЫЫФБл ЃаЃХЃЧ ЖЫЭЗЃЌдкШ§ЙЩТна§НЛСЊзїгУЯТЕУЕНСЫЮяРэЫЎФ§НКЁЃгЩгк ЃаЃЯЃЧЃЙ Ш§ЙЩТна§здзщзАОпгаПЩФцЕФЮТУєадЃЌМДЮТЖШИпгкНтСДЮТЖШЃЈЃдЃэ ЃЉЪБШ§ЙЩТна§НсЙЙБЛЦЦЛЕЃЌЖјЮТЖШЕЭгк ЃдЃэ ЪБШ§ЙЩТна§НсЙЙгжжиаТаЮГЩЃЌЫљвдЛљгкШ§ЙЩТна§ЮЊЮяРэНЛСЊНсЙЙЕФЫЎФ§НКЕФНЛСЊУмЖШКмШнвзЭЈЙ§ЮТЖШРДЕїНкЃЌДгЖјЪЙЕУетжжЫЎФ§НКЕФСІбЇадФмОпгаПЩЕїадЁЃГ§ДЫжЎЭтЃЌМгШыздгЩЕФ ЃУЃЭЃа ПЩВЮгыОКељЫЎФ§НКЬхЯЕжа ЃаЃЯЃЧЃЙ ЕФШ§ЙЩТна§ЕФаЮГЩЃЌЭЌбљвВФмЕїПиЫЎФ§НКЕФСІбЇадФмЁЃВЛЭЌгкЫФБлЕФ ЃаЃХЃЧЃЌЃУЃшЃэЃщЃхЃьЃхЃїЃѓЃыЃщ ПЮЬтзщЃлЃДЃЙЃнЭЈЙ§ТѕПЫЖћМгГЩЗДгІНЋФЃФтЖрыФ ЃаЃЯЃЧЃИ НгжІЕНАЫБл ЃаЃХЃЧ ЕФЖЫЭЗЃЌЭЈЙ§ОпгаЮТЖШПЩФцадШ§ЙЩТна§НјааЮяРэНЛСЊЃЌЭЌбљвВЕНСЫОпгаЮТУєадЕФЮяРэЫЎФ§НКЃЌМДЫЎФ§НКдкЮТЖШСйНќ ЃдЃэ ЪББфЮЊНќЫЦвКЬЌЃЌЖјЫцзХЮТЖШЕЭжСЪвЮТЪБЫќгжжиаТБфЮЊ№ЄЕЏадЕФзДЬЌЃЌШчЭМЪН ЃДЁЃЕБ ЃИ ЃсЃђЃэ ЃаЃХЃЧ⁃ЃаЃЯЃЧЃИ ЕФХЈЖШЗжБ№ЮЊ ЃД ЃїЃєЃЅ КЭ ЃИ ЃїЃєЃЅ ЪБЃЌЦфШмвКПЩдк ЃБ ЃэЃщЃю ФкаЮГЩФ§НКЁЃФ§НКЕФДЂФмФЃСПЗжБ№ЮЊ ЃЖЃИЃЕ ЃаЃс КЭ ЃБЃГЃГЃЗЃаЃсЃЌвђДЫЫЎФ§НКЕФЧПЖШПЩЭЈЙ§ИФБфШмвКГѕЪМХЈЖШРДЕїНкЁЃИУПЮЬтзщгждкДЫбаОПЛљДЁЩЯЃЌНЋВЛЭЌГЄЖШЕФФЃФтЖрыФ ЃаЃЯЃЧЃю ЃЈЃю ЃН ЃЗЁЂЃИЁЂЃЙЃЉНгжІЕНСЫАЫБл ЃаЃХЃЧ ЕФЖЫЭЗЃлЃЕЃАЃнЃЌВЂЭЈЙ§баОПЗЂЯжЫцзХФЃФтЖрыФСДЕФдіГЄЃЌЕЅЖРЕФ ЃаЃЯЃЧЃювдМА ЃаЃХЃЧ⁃ЃаЃЯЃЧЃю аЮГЩЕФШ§ЙЩТна§ЕФШШЮШадЖМЛсУїЯдЕФЬсИпЃЌЯргІЕФ ЃаЃХЃЧ⁃ЃаЃЯЃЧЃю аЮГЩЕФШ§ЙЩТна§ЕФШШЮШЖЈадвВвЊИпгкЕЅЖРЕФ ЃаЃЯЃЧЃю ЃЌ ЧвЫцзХЃаЃЯЃЧЃю СДГЄЕФдіМгЃЌФ§НКЭјТчжаЕФЮяРэНсЙЙБфЕУИќЮШЖЈЃЌЯргІЕФ ЃаЃХЃЧ⁃ЃаЃЯЃЧЃю Ф§НКЕФФ§НК⁃ШмвКзЊБфЮТЖШвВгаЫљЬсИпЁЃбаОПЛЙЗЂЯжЃЌЪвЮТЯТ ЃаЃЯЃЧЃю СДГЄЕФдіМгЖд ЃИ ЃсЃђЃэ ЃаЃХЃЧ⁃ЃаЃЯЃЧЃю Ф§НКЧПЖШЕФгАЯьВЛДѓЃЈЃИ ЃсЃђЃэЃаЃХЃЧ⁃ЃаЃЯЃЧЃю ХЈЖШЮЊ ЃДЃЅ Ѓї ЃЏ Ѓі ЪБЃЌДЂФмФЃСПдк ЃЖЃДЃА ЁЋЃЗЃАЃА ЃаЃс МфБфЛЏЃЉЁЃ
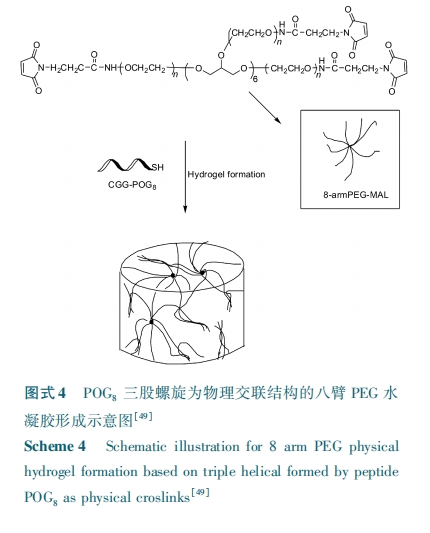
ЩЯЪібаОПЖМЪЧМЏжагкШ§ЙЩТна§ЕФЮТУєадКЭздзщзАадЃЌЖјУЛгабаОПЖрБл ЃаЃХЃЧ ЕФБлЪ§ЖдаЮГЩДѓЗжзгЕФЙЙЯѓКЭНЛСЊаджЪЕФгАЯьЁЃЃЭЃсЃєЃѓЃѕЃѓЃсЃыЃщ ЕШЃлЃЕЃБЃн НЋ ЃаЃЯЃЧЃБЃАаоЪЮЕНСЫЖрБл ЃаЃХЃЧ ЕФЖЫЭЗЩЯЃЈЃю ЃсЃђЃэ ЃаЃХЃЧЃЌ Ѓю ЃН ЃБЃЌЃВЃЌЃДЃЌЃИЃЉЕУЕНСЫвЛЯЕСаЕФГЌЗжзгНсЙЙЃЌЭЈЙ§баОПЗЂЯж ЃюЃсЃђЃэ ЃаЃХЃЧ⁃ЃаЃЯЃЧЃБЃАЕФСїЬхжБОЖЫцзХ Ѓю ЕФдіЖрЯШдіДѓКѓМѕаЁЃЌЦфжа ЃД ЃсЃђЃэ ЃаЃХЃЧ⁃ЃаЃЯЃЧЃБЃАОпгазюДѓСїЬхжБОЖЃЌЧвЮТЖШЖдСїЬхжБОЖвВгавЛЖЈЕФгАЯьЁЃгЩгкШ§ЙЩТна§МфПЩЭЈЙ§ЧтМќЯрЛЅзїгУЃЌЫћУЧНЋ ЃаЃЯЃЧЃБЃА аоЪЮЕФЖрБлЃаЃХЃЧ зїЮЊЂёаЭНКдЕФФЩУзФ§НКвђзгЁЃЭЈЙ§баОПЗЂЯж ЃД ЃсЃђЃэ ЃаЃХЃЧ⁃ЃаЃЯЃЧЃБЃАНЛСЊЕФНКдФ§НКОпгазюДѓЕФДЂФмФЃСПЃЌДгЖјПЩЭЦЖЯ ЃаЃХЃЧ ЕФБлЪ§ШЗЪЕгАЯьЕНСЫЦфЬхЯЕжаШ§ЙЩТна§ЕФУмЖШЃЌНјЖјгАЯьСЫНКдЫЎФ§НКЬхЯЕжаЕФЮяРэНЛСЊУмЖШЁЃбаОПвВЗЂЯж ЃД ЃсЃђЃэ ЃаЃХЃЧ⁃ЃаЃЯЃЧЃБЃАЕФХЈЖШврЖдЦфНЛСЊЕФНКдФ§НКЕФДЂФмФЃСПгаНЯДѓЕФгАЯьЃЌИУФ§НКЕФзюДѓДЂФмФЃСППЩДя ЃВ.ЃЕ ЃыЃаЃсЁЃ
вдЩЯЛљгк ІТ⁃елЕўНсЙЙЁЂОэЧњТна§НсЙЙвдМАШ§ЙЩТна§НсЙЙздзщзАЕУЕНЕФЫЎФ§НКОпгаКмЖргХвьЕФНсЙЙПижЦЬиадЃЌБШШчЭтНчЛЗОГДЬМЄЯьгІадЁЃЭЌЪБДѓВПЗжИУРрОлКЯЮядкгЩШмвКЕНФ§НКзЊБфЕФЙ§ГЬжаЖМашОЙ§вЛЖЈЕФЪБМфЃЌетЮЊЪжЪѕВйзїЬсЙЉСЫБуРћЃЌвђДЫЮДРДИУРрФ§НКПЩЭћЭЈЙ§ЕїНкФ§НКжЦБИВЮЪ§ДгЖјЭЈЙ§дЮЛзЂЩфЕФЗНЪНдкШБЫ№ВПЮЛаЮГЩЫљашЕФШЮвтаЮзДРДЬюВЙШБЫ№Лђеп№ЄКЯЪмЫ№ЕФзщжЏЁЃШЛЖјЃЌИУРрЫЎФ§НКЭјТчЛљБОЖМЪЧЮяРэНЛСЊЫљЕУЃЌСІбЇадФмНЯВюЃЌЦфФ§НКЪБМфМАФ§НКаджЪОпгаУїЯдЕФХЈЖШвдМАБЃЮТЪБМфвРРЕадЃЌФ§НКЕФЭтНчДЬМЄЯьгІФмСІвВгаД§НјвЛВНЬсИпЃЌетаЉвВдквЛЖЈГЬЖШЩЯзшАСЫЦфдкЩњЮявНгУСьгђЕФгІгУЁЃЖрыФЕФДцдкЪЙЕУетРрЮяРэФ§НКдкЬхФквзЕААзЫЎНтЃЌОЁЙмШЫЙЄКЯГЩОлКЯЮяБОЩэвЛЖЈГЬЖШЩЯФмЛКНтЖрыФЕААзЫЎНтЕФЮЪЬтЃЌЕЋетЗНУцбаОПЙЄзїШдУцСйзХжюЖрЕФЬєеНЁЃ
ЃГ ЛЏбЇНЛСЊЫЎФ§НК
ЖрыФздзщзАжїЕМСЫЩЯЪіОлКЯЮяЮяРэФ§НКЕФаЮГЩЃЌВЂЬсЙЉСЫФ§НКвдгХСМЕФНсЙЙПижЦЬиадЁЃШЛЖјвЛАуРДНВЮяРэФ§НКЕФСІбЇадФмДѓЖрНЯШѕЃЌЫљвджкЖрбаОПНЋНЙЕуЙизЂЕНЛЏбЇМќНЛСЊЕФЫЎФ§НКЩЯЁЃЭЌЪБжЕЕУзЂвтЕФЪЧдкЩЯЪіЮяРэФ§НКжаЃЌФГаЉЖрыФвбЦ№ЕНСЫГ§НсЙЙПижЦжЎЭтЕФЬиЪтЙІФмЛЏзїгУЃЌБШШчЛљгк ІТ⁃елЕўЕФЬижжЖрыФзщзАаЮГЩЕФФЩУзЯЫЮЌПЩДйНјєЧЛљСзЛвЪЏПѓЛЏЃлЃГЃДЃнЁЃвђДЫЃЌЯжНёвЛаЉБЈЕРНЋЖрыФв§ШыЕНЛЏбЇНЛСЊЕФФ§НКЭјТчжаЃЌетРяЕФЖрыФжївЊзїгУВЛдкгкЦфЖдФ§НКНсЙЙаЮГЩЕФЙБЯзЃЌЖјЪЧЮЊСЫИГгшдБОЕФЛЏбЇФ§НКвдЩњЮябЇЙІФмЃЌБШШчЯИАћ№ЄИНЁЂУИНЕНтЁЂПЙОњадЕШЁЃ
ЃГ.ЃБ ЯИАћ№ЄИНад
ВЛЭЌадФмЕФШЫЙЄКЯГЩОлКЯЮяДѓЖрОпгазуЙЛЕФСІбЇЮШЖЈадЁЂЕЏадЁЂЮоЖОвдМАПЙНЕНтЮШЖЈадЃЌБЛЙуЗКгІгУгквНгУЗНУцЃЌЕЋЦфКЭЯИАћжЎМфЕФзїгУСІВЛзуЃЌвзЕМжТЬхФкЗЂбзЁЂбЊЫЈаЮГЩЕШВЛСМЗДгІЁЃЮЊСЫНтОіетвЛЮЪЬтЃЌзюГѕжївЊЪЙгУ№ЄИНадЕФЕААзРДаоЪЮВФСЯЕФБэУцРДИФЩЦВФСЯЖдЯИАћЕФ№ЄИНадЃЌЕЋетбљЕФЗНЗЈДцдкжюЖрШБЕуЃЌБШШчвзЕААзНЕНтЁЂУтвпдадЃлЃЕЃВЃнЁЃдкКѓРДбаОПжаЗЂЯжетаЉ№ЄИНадЕААзЕФЯИАћ№ЄИНЮЛЕуНігЩЪ§ИіАБЛљЫсзщГЩЃЌЦфжабаОПБШНЯЩюШыЁЂгІгУБШНЯЙуЗКЕФЪЧКЌ ЃвЃЧЃФ ађСаЖрыФЃЌгЩгкетжжШ§ыФФмгыЯИАћБэУцЕФећСЊЕААзЪмЬхНсКЯЃЌДгЖјПЩДйНјЯИАћдкВФСЯБэУцЕФЮќИНЁЃКЌ ЃвЃЧЃФ ађСаЕФЖрыФАќРЈСНжжЃЌКЌ ЃвЃЧЃФ ађСаЕФЯпадЖрыФКЭКЌ ЃвЃЧЃФ ађСаЕФЛЗаЮЖрыФЃЌЯТУцНіЖдКЌЃвЃЧЃФ ЕФЯпадЖрыФзїМђвЊЕФзмНс.
дкЙ§ШЅЕФбаОПжаЃЌЮЊСЫИФЩЦЯИАћдкЫЎФ§НКЭјТчжаЕФ№ЄИНЁЂРЉЩЂвдМАдіжГадЃЌКЌ ЃвЃЧЃФ ађСаЖрыФвбБЛв§ШыЕНЭИУїжЪЫсЃлЃЕЃГ ЃЌЃЕЃДЃнЁЂ ОлввЯЉДМ ЃЈ ЃаЃжЃСЃЉЃлЃЕЃЕЃн вдМАЃаЃХЃЧЃлЃЕЃЖ ЁЋ ЃЕЃЙЃнЕШЫЎФ§НКЕФЬхЯЕжаЁЃв§ШыЕФЗНЗЈжївЊгаСНжжЃКвЛЪЧ ЃвЃЧЃФ ЫцзХНЛСЊМСв§ШыЕНЛЏбЇЭјТчНсЙЙжаЃЌБШШч ЃгЃшЃѕ ЕШЃлЃЕЃГЃнРћгУ ЃУЃУЃвЃЧЃФЃг ЖрыФађСажаЫЋлЯЛљКЭОлввЖўДМЖўБћЯЉЫсѕЅЃЈЃаЃХЃЧЃФЃСЃЉЕФТѕПЫЖћМгГЩЗДгІЃЌЕУЕНСЫЗжзгЖЫЭЗКЌгаЫЋМќЃЌжаМфКЌга ЃвЃЧЃФ ЕФЃаЃХЃЧЃФЃС⁃ЃвЃЧЃФ⁃ЃаЃХЃЧЃФЃС НЛСЊМСЃЌдйКЭлЯЛљаоЪЮЭИУїжЪЫсМфЕФТѕПЫЖћМгГЩЗДгІНЋ ЃвЃЧЃФ в§ШыЕНЫЎФ§НКЕФЛЏбЇЭјТчжаЃЛЖўЪЧЯШНЋ ЃвЃЧЃФ зїЮЊжЇСДВПЗжНгжІЕНОлКЯЮяЕФжїСДЩЯЃЌШЛКѓ ЃвЃЧЃФ ЫцзХОлКЯЮяЕФНЛСЊБЛв§ШыЕНЛЏбЇЭјТчжаЃЌБШШч ЃгЃуЃшЃэЃхЃфЃьЃхЃю ЕШЃлЃЕЃЕЃн ЗжБ№НЋЫЋМќЛљЭХКЭ ЃвЃЧЃФ ЭЈЙ§ЛЏбЇаоЪЮМоНгЕНСЫ ЃаЃжЃС ЕФВрСДЩЯЃЌзюКѓЙтНЛСЊЕУЕНСЫОпгаЯИАћ№ЄИНадЕФЫЎФ§НКЁЃГ§ДЫжЎЭтЃЌЫЎФ§НКШ§ЮЌЭјТчЬхЯЕжа ЃвЃЧЃФ ЖдЯИАћЕФдіжГвдМАзщжЏЕФЩњГЩгАЯьОпгаХЈЖШвРРЕадЃлЃЖЃАЃнЁЃ
ЃзЃсЃюЃч ЕШЃлЃЖЃБЃнЯШгУРвАБЫсЃЈЃдЃљЃђЃсЃэЃщЃюЃхЃЌЃдЃљЃђЃЉЖдЭИУїжЪЫсЃЈЃШЃСЃЉЛЏбЇИФадЕУЕНСЫ ЃШЃС⁃ЃдЃљЃђЃЌгжНЋ ЃвЃЧЃФ КЭЖдєЧЛљБНБћЫсЖд ЃаЃХЃЧ ЕФСНЖЫаоЪЮЕУЕНСЫЭЌЪБКЌЫЋБНЗгЛљКЭ ЃвЃЧЃФ ЕФ ЃаЃХЃЧ ЗжзгЃЌдкРБИљЙ§бѕЛЏУИ ЃШЃвЃа КЭЙ§бѕЛЏЧтЃЈЃШЃВЃЯЃВ ЃЉЕФзїгУЯТСНепМфЕФБНЗгЛљЃЈЃ№ЃшЃхЃюЃяЃь ЃэЃяЃщЃхЃєЃљЃЉЗДгІЕУЕНСЫ ЃШЃС⁃ЃдЃљЃђ⁃ЃвЃЧЃФ дЮЛФ§НКЃЌШчЭМЪН ЃЕЁЃЭЈЙ§ЖдШЫЦъОВТіФкЦЄЯИАћХрбјЗЂЯжЃЌО ЃвЃЧЃФ ЖрыФЛЏбЇаоЪЮЕФ ЃШЃС⁃ЃдЃљЃђ ЫЎФ§НКБШУЛгааоЪЮЕФ ЃШЃС⁃ЃдЃљЃђ ЫЎФ§НКОпгаИќЧПЕФЯИАћ№ЄИНадЃЌДгЖјИФЩЦСЫЯИАћдкЫЎФ§НКжаЕФдіжГЁЂЧЈвЦвдМАУЋЯИбЊЙмзДЭјТчНсЙЙЕФаЮГЩЁЃ
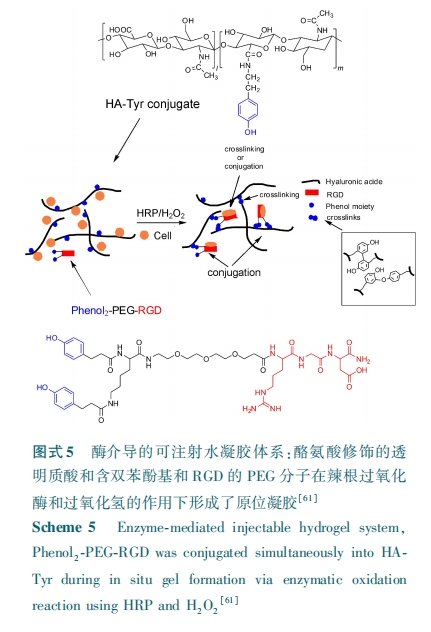
ЃГ.ЃВ УИНЕНтад
жЇМмВФСЯНЕНтЫйТЪЪЧжВШыВФСЯашвЊПМТЧЕФзюживЊЮЪЬтжЎвЛЃЌЯЃЭћЦфНЕНтЫйТЪгызщжЏШБЯнЮЛжУаТзщжЏдйЩњЕФЫйТЪЯрЦЅХфЃЌШчЙћЦфНЕНтЫйТЪЯрЖдгкзщжЏдйЩњЫйТЪЙ§ПьЃЌОЭЪЇШЅСЫЫќЖдЯИАћЩњГЄЕФдиЬхЙІФмЁЃЗДжЎЃЌОЭЛсзшАаТзщжЏЕФдйЩњЁЃЖјДѓЖрЪ§ПЩНЕНтКЯГЩОлКЯЮяЃЌБШШчОлМКФкѕЅЁЂОлШщЫсЁЂОлввНЛѕЅЕФНЕНтжївЊЪЧЛљгкжїСДѕЅЕФЫцЛњЫЎНтЃЌетаЉЫЎНтЙ§ГЬЖдЯИАћаХКХвдМАЯИАћЗжУкЕФУИВЂВЛОпгаЯьгІадЁЃЮЊСЫИГгшжЇМмВФСЯЩњЮяНЕНтадЃЌЦфжазюКУЕФЗНЗЈОЭЪЧРћгУЯИАћЭтЛљжЪЕААзЫЎНтНЕНтЛњжЦЃЌНЋЖдУИОпгаЯьгІадЕФЖрыФађСав§ШыЕНжЇМмВФСЯжаЃлЃЖЃВЃнЁЃ
Лљ жЪ Н№ Ъє ЕА Аз УИ ЃЈ ЃэЃсЃєЃђЃщЃј ЃэЃхЃєЃсЃьЃьЃяЃ№ЃђЃяЃєЃхЃщЃюЃсЃѓЃхЃѓЃЌЃЭЃЭЃаЃѓЃЉЪЧвЛРрыФСДФкЧаУИЃЌЖдЯИАћЭтЛљжЪзщЗжЕФНЕНтЦ№зХживЊЕФзїгУЁЃЮЊСЫИГгшЫЎФ§НКЕФУИНЕНтадЃЌгабаОПепЗжБ№НЋЖдЛљжЪН№ЪєЕААзУИ ЃБЃЈЃЭЃЭЃаЃБЃЉКЭЛљжЪН№ЪєЕААзУИ ЃВЃЈЃЭЃЭЃаЃВЃЉгаЯьгІадЕФвЛЯЕСаЖрыФађСазїЮЊНЛСЊМСЕФВПЗжв§ШыЕНСЫ ЃаЃХЃЧ ЫЎФ§НКЕФЬхЯЕжаЃлЃЖЃГЃнЁЃвВгабаОПепНЋЖдЛљжЪН№ЪєЕААзУИ ЃВЃЈЃЭЃЭЃаЃВЃЉОпгаЯьгІадЕФ ЃЧЃаЃбЃЧЁ§ЃЩЃСЃгЃб КЭ ЃаЃжЃЧЁ§ЃЬЃЩЃЧЃЈЁ§БэЪОЖЯСбЕуЃЉЖрыФађСавВЗжБ№зїЮЊНЛСЊМСЕФВПЗжв§ШыЕНСЫЦЯОлЬЧЃлЃЖЃДЃн КЭдхЫсбЮЃлЃЖЃЕЃн ЫЎФ§НКЬхЯЕжаЁЃРћгУЭЌбљЕФдРэЃЌбаОПепНЋЖдЛљжЪН№ЪєЕААзУИ ЃБЃГЃЈЃЭЃЭЃаЃБЃГЃЉОпгаЯьгІадЕФ ЃаЃбЃЧЁ§ЃЬЃСЃЫ ЖрыФађСав§ШыЕНСЫОл ЃЮ⁃вьБћЛљБћЯЉѕЃАЗгыБћЯЉЫсЙВОлЮяЫЎФ§НКЬхЯЕжаЃлЃЖЃЖЃнЁЃЙтНЛСЊЕФМзЛљБћЯЉЫсЛЏЕФЭИУїжЪЫсЫЎФ§НКЫфПЩзїЮЊШЫЙЧ Ыш Мф Гф жЪ ИЩ ЯИ Аћ ЃЈ ЃшЃѕЃэЃсЃю ЃэЃхЃѓЃхЃюЃуЃшЃљЃэЃсЃь ЃѓЃєЃхЃэЃуЃхЃьЃьЃѓЃЌ ЃшЃЭЃгЃУЃѓЃЉЕФШэЙЧаЮГЩжЇМмВФСЯЃЌЕЋФ§НКЭјТчНсЙЙжаЕФЛЏбЇНЛСЊШДВЛФмНЕНтЃЌДгЖјзшАСЫШэЙЧЛљжЪЕФПеМфЗжВМвдМАЯИАћЗжЩЂЁЃЃЦЃхЃюЃч ЕШЃлЃЖЃЗЃн ЗжБ№НЋЖдЃЭЃЭЃа УєИаЕФ ЃЧЃУЃвЃФЃжЃаЃЭЃгЁ§ЃЭЃвЃЧЃЧЃФЃвЃУЃЧ ЖрыФКЭЖдЃЭЃЭЃа ВЛУєИаЕФЖўСђЫеЬЧДМЃЈЃФЃФЃдЃЉзїЮЊТэРДѕЃбЧАЗЃЈЃэЃсЃьЃхЃщЃэЃщЃфЃхЃЌ ЃЭЃСЃЉЛЏЕФЭИУїжЪЫсЃЈЃШЃСЃЉЕФНЛСЊМСЃЌДгЖјЕУЕНСЫСНжжаджЪВЛЭЌЕФЫЎФ§НКЃЈЃЭЃСЃШЃС⁃ЃЭЃЭЃа КЭЃЭЃСЃШЃС⁃ЃФЃФЃдЃЉЃЌзїепНЋ ЃшЃЭЃгЃУЃѓ ЗтзАдкСНжжЫЎФ§НКжаНјааЯИАћХрбјЃЌШчЭМЪН ЃЖЁЃбаОПБэУї ЃЭЃСЃШЃС⁃ЃЭЃЭЃа ЫЎФ§НКжаЕФ ЃшЃЭЃгЃУЃѓ ФмЙЛжиЫмЦфжмЮЇЕФЫЎФ§НКвдМАдкФ§НКжаФмНјааЧЈвЦЃЌетжжЯИАћНщЕМЕФФ§НКНЕНтФмДйНј ЃшЃЭЃгЃУЃѓ ЕФШэЙЧаЮГЩвдМАФмвжжЦ ЃшЃЭЃгЃУЃѓ Й§ЖШЩњГЄЁЃОЙ§ГЄЦкЯИАћХрбјЗЂЯж ЃЭЃСЃШЃС⁃ЃЭЃЭЃа ЯрБШгк ЃЭЃСЃШЃС⁃ЃФЃФЃд Ф§НКжаГСЛ§СЫИќЖрЕФШэЙЧЛљжЪЃЌМѕЩйСЫИЦЛЏЕФЗЂЩњЁЃГ§СЫЛљжЪН№ЪєЕААзУИЭтЃЌвВгабаОПепНЋЖдЯЫШмУИКЭЕЏадЕААзУИОпгаЯьгІадЕФЖрыФађСав§ШыЕНСЫЫЎФ§НКЬхЯЕжаЃлЃЖЃИЃЌ ЃЖЃЙЃнЁЃ
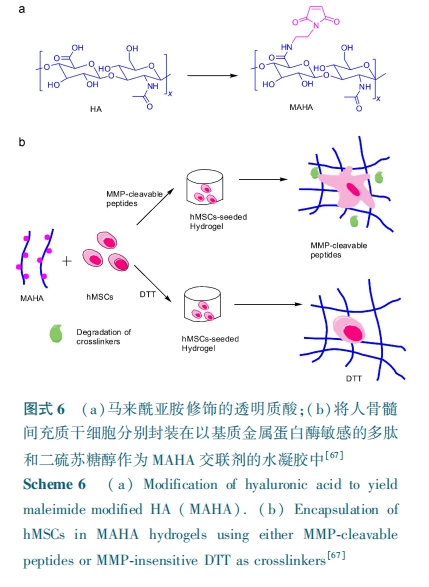
ЃГ.ЃГ ПЙОњад
зюНќМИФъгабаОПепБЈЕРСЫНЋЖрыФЕФПЙОњадв§ШыЕНЛЏбЇНЛСЊЕФОлКЯЮяжаЃЌДгЖјИГгшЫЎФ§НКвдПЙОњадЁЃЃкЃшЃяЃѕ ЕШЃлЃЗЃАЃнЭЈЙ§ ІХ⁃ОлРЕАБЫсЃЈЃХЃаЃЬЃЉЖЫЭЗАБЛљКЭМзЛљБћЯЉЫсЃЈЃЭЃСЃЉєШЛљМфЕФѕЃАЗЛЏЗДгІЕУЕНСЫ ЃХЃаЃЬ⁃ЃЭЃСЃЌШЛКѓНЋЦфКЭ ЃаЃХЃЧЃФЃС вдМА ЃЮЃЌ ЃЮ⁃ЖўМзЛљБћЯЉѕЃАЗШ§епдкЙтНЛСЊзїгУЯТЕУЕНСЫЫЎФ§НКЃЌгЩгкбєРызгЕФЃХЃаЃЬ ПЩЮќИНдкЩњЮяФЄЕФБэУцЕМжТЯИАћЕФЩњРэЫ№ЩЫЃЌЪЙЕУФ§НКОпгаЙуЦзЕФПЙОњадЁЃЃгЃяЃюЃч ЕШЃлЃЗЃБЃн КЯГЩСЫвЛЯЕСаЕФ Ѓ№ЃяЃьЃљЃЈ ЃЬЃљЃѓЃЉЃј ЃЈЃСЃьЃсЃЉЃљ ЃЈ Ѓј ЃЋ Ѓљ ЃН ЃБЃАЃАЃЌ ЃЬЃљЃѓЃКРЕАБЫсЃЌЃСЃьЃсЃКБћАБЫсЃЉЃЌ ЦфНсЙЙжаРЕАБЫсВаЛљВЛНіПЩзїЮЊСљБлОлввЖўДМѕЃАЗЛљчњчъѕЃбЧАЗЬМЫсѕЅ ЃЈ ЃЖ ЃсЃђЃэЃаЃХЃЧ⁃ЃСЃгЃЧЃЉ ЕФНЛСЊМСЃЌЭЌЪБРЕАБЫсВаЛљЕФДцдквВИГгшСЫЫЎ Ф§ НК вд ПЙ Оњ адЁЃНј вЛ ВН ба ОП ЗЂ Яж Ѓ№ЃяЃьЃљЃЈЃЬЃљЃѓЃЉЃЖЃА ЃЈЃСЃьЃсЃЉЃДЃА КЭЃЖ ЃсЃђЃэ ЃаЃХЃЧ⁃ЃСЃгЃЧ НЛСЊЕУЕНЕФЫЎФ§НКЭЌЪБОпгаПЙОњадКЭЯИАћ№ЄИНадЃЌЪЙЦфПЩзїЮЊЧБдкЕФЦЄЗєДДЩЫгњКЯжЇМмВФСЯЁЃГ§СЫОлРЕАБЫсЭтЃЌЛЙгабаОПепНЋЦфЫћОпгаПЙОњадЕФЖрыФв§ШыЕНОлКЯЮяжаЃЌЃУЃьЃхЃяЃ№ЃшЃсЃѓ ЕШЃлЃЗЃВ ЃЌЃЗЃГЃн НЋ КЌ га Аы ыз АБ Ыс ЕФ ПЙ Оњ ыФЃЈЃУЃЫЃвЃзЃзЃЫЃзЃЩЃвЃзЃЉЁЂМОЮьЫФДМЫФ⁃ЃГ⁃лЯЛљБћЫсѕЅвдМАЃаЃХЃЧЃФЃС Ш§епЕФЛьКЯвКзЊвЦЕНОлЖдБНЖўМзЫсввЖўДМѕЅЦЌБэУцЃЌШЛКѓЭЈЙ§СђДМ⁃ЯЉЙтОлКЯЗДгІЕУЕНСЫОпгаПЙОњадФмЕФЫЎФ§НКЁЃ
ЃГ.ЃД зщжЏ№ЄИНад
ІХ⁃ОлРЕАБЫсзїЮЊвЛжжЬьШЛЖрыФЃЌвђЦфгызщжЏМфЕФРызгОВЕчЯрЛЅЮќв§зїгУЖјОпгаСМКУЕФзщжЏ№ЄИНадЁЃБО ПЮ Ьт зщ Рћ гУ Тэ РД ѕЃ бЧ АЗ ЛЏ ЕФ Ол РЕ АБ ЫсЃЈЃХЃаЃЬЃЭЃЉКЭлЯЛљЛЏПЧОлЬЧЃЈЃУЃгЃгЃЉМфЕФТѕПЫЖћМгГЩЗДгІЕУЕНСЫ№ЄКЯадСМКУЕФдЮЛЫЎФ§НК№ЄКЯВФСЯЃЌШчЭМЪН ЃЗЁЃЭЈЙ§№ЄКЯСІВтЪдЗЂЯжИУЫЎФ§НКзюИп№ЄКЯСІЃЈЃИЃЗ. ЃЕ ЃыЃаЃсЃЉЪЧЩЬвЕЯЫЮЌЕААзНКЕФ ЃД БЖЃЌетжжСМКУЕФ№ЄКЯСІжївЊРДздОлРЕАБЫсКЭзщжЏМфЕФОВЕчЮќв§зїгУвдМАФ§НКЬхЯЕжалЯЛљКЭТэРДѕЃбЧАЗЛљЭХгызщжЏжаАыызАБЫсИЛМЏзггђаЮГЩЕФЛЏбЇМќКЯзїгУЁЃИљОнлЯЛљЛЏПЧОлЬЧХЈЖШЁЂТэРДѕЃбЧАЗЛљЭХШЁДњЖШвдМАТэРДѕЃбЧАЗЛљгылЯЛљМфЕФФІЖћБШВЛЭЌЃЌИУЬхЯЕЕФФ§НКЪБМфдкЃБЃЕ ЁЋ ЃВЃБЃЕ Ѓѓ жЎМфЁЃКѓРДбаОПжаЗЂЯж ЃаЃХЃЧ ЕФв§ШыПЩИФЩЦФ§НКЕФДЂФмФЃСПЃЌЦфжазюМбФ§НКЬхЯЕЕФДЂФмФЃСППЩДя ЃБЃЖЃБЃД ЃаЃсЃЌВЂЧв ЃаЃХЃЧ СДЖЮСМКУЕФСщЛюадвдМАОљдШЗжЩЂадвВИФЩЦСЫФ§НКЕФФкОлСІЃЌДгЖјЬсИпСЫФ§НКЕФзюже№ЄКЯСІЃЌзюИпПЩДя ЃБЃДЃИ ЃыЃаЃсЃЌЪЧЩЬвЕЯЫЮЌЕААзНКЕФ ЃЖ БЖЃлЃЗЃД ЃЌЃЗЃЕЃнЁЃживЊЕФЪЧЃЌДЫРрЖрЬЧЖрыФЫЎФ§НКПЩЭЈЙ§зЂЩфЕФЗНЪНдкЬхФкдЮЛаЮГЩЃЌетбљОЭБмУтСЫЭтПЦЪжЪѕЙ§ГЬжаЕФИпЖШДДЩЫад ЃЌДгЖјНЕЕЭСЫвНСЦЗбгУКЭМѕЩйЛМепЭДПрЃЌВЂЧвЦфдСЯРДдДЗсИЛЁЂПЩЩњЮяНЕНтЁЂЩњЮяЯрШнадСМКУЃЌЪЙЦфдкЩњЮявНвЉСьгђгазХОоДѓЕФгІгУЧБСІЁЃ
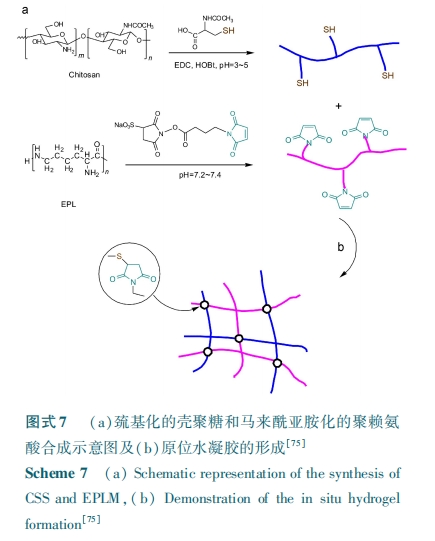
жЕЕУзЂвтЕФЪЧЛЏбЇМќНЛСЊЕФЫЎФ§НККмЖрВЩгУСЫЙтНЛСЊММЪѕЃЌзїЮЊгІгУгкЩњЮявНгУСьгђЕФЫЎФ§НКВФСЯЃЌВаСєЕЅЬхЕФШЅГ§вдМАв§ЗЂМСЛђЙтУєМСЕФЪЙгУЪЧашвЊИјгшЙизЂЕФЮЪЬтЁЃФПЧАДѓЖрЪ§ЬхЯЕЪЙгУЕФЙтв§ЗЂМС ЃЩЃђЃчЃсЃуЃѕЃђЃх ЃВЃЙЃЕЃЙ БЛШЯЮЊЪЧвЛжжЩњЮяЯрШнадНЯКУЕФв§ЗЂМСЃлЃЕЃЕЃЌЃЗЃАЃЌЃЗЃЖЃЌЃЗЃЗЃнЃЌЬхЭтЯИАћХрбјЪЕбщжЄУїЕЭМССПЃЩЃђЃчЃсЃуЃѕЃђЃх ЃВЃЙЃЕЃЙ в§ЗЂНЛСЊЕФЫЎФ§НКЖдВИШщЖЏЮяЯИАћВЂУЛгаЖОадЃлЃЗЃЗЃнЁЃЭЌЪБЃЌвВгаБЈЕРРћгУгаЛњШмМСнЭШЁЁЂРыаФЗжРыГСЕэЕФЗНЗЈРДШЅГ§ВаСєЕЅЬхЃлЃГЃИЃнЃЌвдМАЖдНЛСЊЕФФ§НКНјааНўХнГхЯДЕФЗНЗЈвдОЁПЩФмШЅГ§ВаСєЕФаЁЗжзгЮяжЪЃлЃЗЃИЃнЁЃ
ЃД ЮяРэЃЏ ЛЏбЇЫЋжиНЛСЊЫЎФ§НК
ЯрЖдгкЩЯЪіЕЅвЛЮяРэНЛСЊФ§НККЭЛЏбЇНЛСЊФ§НКЃЌНќФъРДГіЯжСЫКЌЖрыФздзщзАЕФЮяРэЃЏ ЛЏбЇЫЋжиНЛСЊЫЎФ§НКЁЃдкетРрЕФЬхЯЕжаЃЌЖрыФВЛЕЋВЮгыСЫФ§НКЭјТчЕФЙЙНЈКЭЖдЭјТчНсЙЙЕФПижЦЃЌЖјЧвЛЙИГгшСЫЫЎФ§НКФГаЉЙІФмЃЌШчУИНЕНтКЭДйзщжЏаоИДЕШЁЃзюживЊЕФЪЧЖрыФздзщзАЙБЯзЕФЮяРэНЛСЊНсЙЙЛЙФмЖдЛЏбЇНЛСЊФ§НКЭјТчЦ№ЕНдіЧПзїгУЁЃЖрыФЕФв§ШыШЗЪЕИјДЫРрФ§НКДјРДСЫжюЖргХСМЬиадЃЌШЛЖјетРрЫЎФ§НКЬхЯЕжаашвЊзЂвтЃКЮЊГфЗжЗЂЛгЖрыФЕФЮяРэадФмЃЌв§ШыЕФЛЏбЇНЛСЊВЛФмЙ§ЖШИЩШХЬхЯЕжаЕФЖрыФздзщзАЁЃ
ЛљгкШ§ЙЩТна§НсЙЙЕФЮТУєЬиадЃЌвВгабЇепНЋШ§ЙЩТна§НсЙЙзїЮЊЮяРэНЛСЊв§ШыЕНСЫЛЏбЇНЛСЊЕФЃаЃХЃЧ ЛљЫЎФ§НКЕФЬхЯЕжаЃЌДгЖјРћгУШ§ЙЩТна§ЕФПЩФцадЗНБуЕиЕїПиШ§ЮЌжЇМмВФСЯЕФСІбЇКЭЩњЮяЛЏбЇадФмЁЃЃйЃѕ ПЮЬтзщЃлЃЗЃЙЃнНЋНКдФЃФтЖрыФ ЃаЃЯЃЧЃЗ НгжІЕНСЫБћЯЉЫс⁃Ол вв Жў ДМ⁃єЧ Лљ чњ чъ ѕЃ бЧ АЗ ЃЈ ЃСЃУЃвЃЬ⁃ЃаЃХЃЧ⁃ЃЮЃШЃгЃЉжїСДЕФвЛЖЫЃЌЕУЕНСЫСНЖЫЭЗЗжБ№ЮЊНКдФЃФтЖрыФ ЃаЃЯЃЧЃЗ КЭЫЋМќЕФ ЃаЃХЃЧ ДѓЗжзгЃЌВЂКЭ ЃаЃХЃЧЃФЃС ЙтНЛСЊЕУЕНСЫвЛжжЫЎФ§НКЁЃЦфжаФЃФтЖрыФ ЃаЃЯЃЧЃЗ аЮГЩЕФШ§ЙЩТна§НсЙЙдкЫЎФ§НКЬхЯЕжаЦ№ЕНСЫЮяРэНЛСЊЕФзїгУЃЌетаЉЮяРэНЛСЊНсЙЙПЩвдБЛЯИАћЗжУкЕФЕААзУИНјааНЕНтЃЌДгЖјПЩвЦГ§ЛЏбЇНЛСЊЭјТчжаЕФШ§ЙЩТна§НсЙЙЖјВњЩњПЊПзЃЌЪЙЕУФ§НКИќЪЪгІЯИАћЕФдіжГКЭЧЈвЦЃЌЧвФЃФтЖрыФ ЃаЃЯЃЧЃЗ КЭНКджЎМфОпгаСМКУЕФЧзКЯадЃЌЪЙЕУетжжЫЎФ§НКФмИќКУЕФБЃСєЯИАћЗжУкЕФНКдЕААзЃЌДйНјСЫзщжЏЕФдйЩњЁЃИУПЮЬтзщНјвЛВННЋМфГфжЪИЩЯИАћЗтзАдкИУЫЎФ§НКжаХрбјЃЌбаОПБэУїИУЫЎФ§НКПЩЮЊМфГфжЪИЩЯИАћЬсЙЉСМКУЕФЮЂЛЗОГЃЌПЩМћетжжЫЎФ§НКПЩгУзїОпгаЩњЮяЛюадЕФШ§ЮЌзщжЏжЇМмЃлЃИЃАЃнЁЃИУПЮЬтзщЃлЃДЃИЃнЛЙНЋЖЫЭЗБЛєЧЛљчњчъѕЃбЧАЗЃЈЃЮЃШЃгЃЉаоЪЮЕФЫФБлОлввЖўДМЃЈЃаЃХЃЧ⁃ЃЮЃШЃгЃЉКЭСНЭЗДјАБЛљЕФ ЃаЃЯЃЧЃЙЖрыФАДФІЖћБШ ЃБЁУ ЃГЕФБШР§ЛьКЯЃЌОЙ§ЛЏбЇЗДгІЕУЕНСЫЭЌЪБКЌгаЮяРэКЭЛЏбЇНЛСЊЕФЫЎФ§НКЃЈЃаЃХЃЧ⁃ЃУЃЭЃаЃЙ⁃ЃЫЃЉЃЌШчЭМЪН ЃИЃсЁЃдкЗДгІЙ§ГЬжаСНЖЫЭЗАБЛљЕФ ЃаЃЯЃЧЃЙЖрыФВЛНіЦ№ЕНСЫЛЏбЇНЛСЊМСЕФзїгУЃЌЭЌЪБгжПЩаЮГЩШ§ЙЩТна§ЃЌвђЖјЯђЫЎФ§НКЬхЯЕжав§ШыСЫЮяРэНЛСЊНсЙЙЁЃЮЊСЫбщжЄетжжЮяРэНЛСЊЕФДцдкдіЧПСЫ ЃаЃХЃЧ ЫЎФ§НКЕФСІбЇадФмЃЌзїепгжНЋЖЫЭЗЭЌбљДјгаАБЛљЕФЃаЃЯЃЧЃЙ ЖрыФКЭЫФБл ЃаЃХЃЧ АДЩЯЪіЕФБШР§ЛьКЯЃЌгЩгкв§Шы ЃаЃЯЃЧЃЙ ЖрыФЕФАБЛљЫсађСаДђТвЃЌДгЖјзшжЙСЫШ§ЙЩТна§ЕФаЮГЩЃЌ ЪЙЕУЕНЕФЫЎФ§НКжЛКЌгаЛЏбЇНЛСЊЃЈЃаЃХЃЧ⁃ЃвЃУЃЭЃаЃЙ⁃ЃЫЃЉЃЌШчЭМЪН ЃИЃсЁЃЖдСНжжЫЎФ§НКЕФВтЪдБэУїетжжШ§ЙЩТна§НсЙЙЙБЯзЕФЮяРэНЛСЊЕФДцдкШЗЪЕЬсИпСЫЫЎФ§НКЕФСІбЇЧПЖШЃЌШчЭМЪН ЃИЃтЁЃЭЈЙ§МгШыздгЩЕФ ЃаЃЯЃЧЃЙ ОКељФ§НКЭјТчжаШ§ЙЩТна§ЕФаЮГЩПЩЕїПиЫЎФ§НКгВЖШЃЌетЪЙЦфдкгУгкЯИАћдіжГКЭЧЈвЦЕФШ§ЮЌжЇМмВФСЯЗНУцгазХЧБдкЕФгІгУЧАОАЁЃ
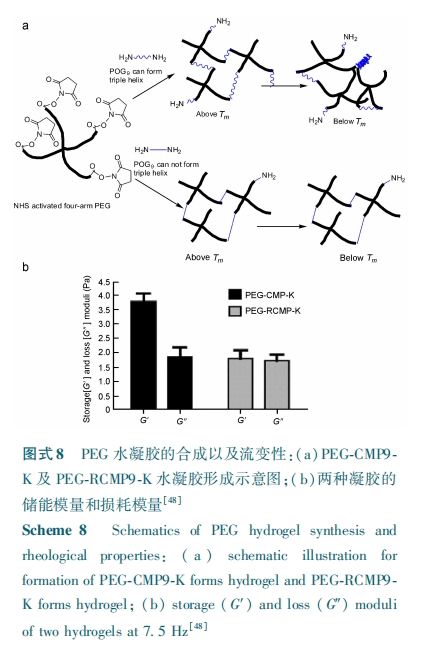
ФПЧАгУгкзщжЏЙЄГЬбаОПЕФЙтНЛСЊЫЎФ§НКЕФЭјТчНсЙЙЪЧЛљгкгРОУСДНгРДЮШЖЈЕФЃЌЮЊСЫЪЙЛЏбЇНЛСЊЕФЫЎФ§НКИќгаРћгкЯИАћЕФЧЈвЦКЭзщжЏЩњГЩЃЌЙ§ШЅжївЊЪЧРћгУЕААзУИСбНтЛђЫЎНтЪЙЕУЛЏбЇФ§НКНЕНтЃЌЕЋетРрЗНЗЈДцдквЛЖЈЕФШБЕуЃЌБШШчНЕНтЫйТЪФбгкПижЦЁЃВЛЭЌгквдЭљЕФНЕНтЛњРэЃЌЃЬЃщЃѕ ЕШНЋОэЧњТна§зїЮЊЮяРэНЛСЊНсЙЙРДЮШЖЈЙтНЛСЊЕФЫЎФ§НКЃлЃЗЃЖЃнЃЌЫћУЧЭЈЙ§вЛЖЮЪшЫЎОлКЯЮяНЋССАБЫсРСДгђЛЏбЇаоЪЮЕН ЃаЃХЃЧЃФЃСвЛЖЫЁЃгЩгкССАБЫсРСДгђПЩзщзАГЩЫФОлЕФОэЧњТна§ЖјЕУЕНСЫЖЫЭЗДјгаЫЋМќЕФЫФБлДѓЗжзгЃЌШЛКѓЙтНЛСЊаЮГЩСЫЫЎФ§НКЃЌШчЭМЪН ЃЙЁЃЪЕбщЗЂЯжИУРрФ§НКЕФДЂФмФЃСПгыЦфХЈЖШгаКмДѓЕФЙиЯЕЃЌМДХЈЖШЮЊ ЃЖЃЅ Ѓї ЃЏ ЃіКЭ ЃБЃАЃЅ Ѓї ЃЏ Ѓі ЪБЃЌ ЦфФ§НКДЂФмФЃСПЗжБ№ЮЊ ЃВЃАЃАЃА ЃаЃс КЭЃЗЃАЃАЃА ЃаЃсЁЃЕБМгШывЛЖЈСПФђЫиЪБЃЌгЩгкССАБЫсРСДгђЕФБфадЪЙЮяРэзщзАЗжНтЖјЦЦЛЕСЫШ§ЮЌЭјТчЃЌжТЪЙЫЎФ§НКЗжНтЮЊШмвКЁЃзїЮЊЖдБШбљЃЌССАБЫсРСДБЛМКЖўЫсЛЏбЇНЛСЊЕФЧЖЖЮЙВОлЮяЃЌФђЫиЕФМгШыФ§НКВЂУЛгаЗжНтЁЃОэЧњТна§здзщзАЪЙЫЎФ§НКФмПЩФцПЊЙиШ§ЮЌЯИАћЧЈвЦЭООЖЃЌЭЌЪБЛЙОпгаСМКУЕФЩњЮяЯрШнадЁЃРћгУЭЌбљЩшМЦЗНЗЈЃЌЃйЃсЃя ЕШЃлЃЗЃЗЃн жЛЪЧИФБфСЫвЛЯТ ЃаЃХЃЧЃФЃСЖЫЭЗЕФЖрыФЃЌКЭЩЯЪіЕФЯжЯѓвЛбљЃЌЕУЕНЕФЫЎФ§НКЖдФђЫиШдгаЯьгІадЁЃДЫЭтЃЌЛљгк ІС⁃Тна§здзщзАЖЏЬЌЕФЖрОлЬхзїЮЊЮяРэНЛСЊНсЙЙЪЙЕУФ§НКОпгаПьЫйздЮваоИДФмСІЃЌДгЖјПЩЪЙВФСЯЗНБуЕиЭЈЙ§здзщзАКЭздЯТЖјЩЯЕФЙЙдьРДЙЙНЈзщжЏЁЃСэЭтЃЌетжжЮяРэЫЎФ§НКЕФжївЊГЩЗжЮЊЩњЮяКЯГЩЕФЖрыФЃЌвђЖјПЩзїЮЊШЫЙЄжЇМмВФСЯРДФЃФтЬьШЛЕФЯИАћЭтЛљжЪЁЃ
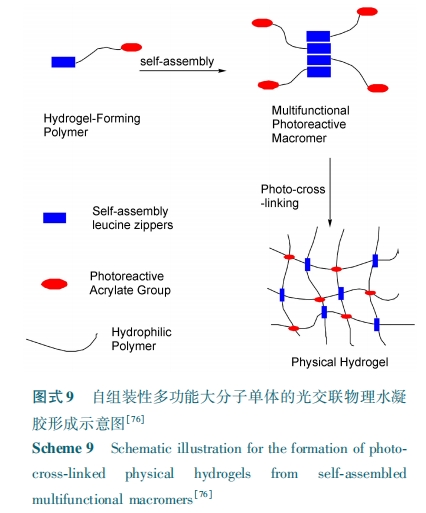
дкетРрЮяРэЃЏ ЛЏбЇИДКЯФ§НКжаЃЌЛљгкЖрыФздзщзАаЮГЩЕФЮяРэНЛСЊНсЙЙВЛНіИГгшСЫЫЎФ§НКЬиЪтЙІФмадЃЌЖјЧвЛЙПЩЬсИпЛЏбЇНЛСЊФ§НКЕФСІбЇадФмЁЃЕЋгЩгкЛЏбЇНЛСЊЭјТчЕФЪјИПВЛРћгкЖрыФЕФздзщзАЃЌЪЙЕУЯрЙиЕФЙЄзїПЊеЙЕУЛЙКмЩйЃЌПМТЧЕНетРрФ§НКзлКЯадФмКЭЙІФмЕФЬсЩ§ЃЌЮДРДдЄЦкетЗНУцЕФЙЄзїЛсгаИќГЄдЖЗЂеЙЁЃ
ЃЕ НсТл
ЖрыФЗжзггЩгкОпгааэЖргХСМадФмЃЌБШШчЩњЮяЯрШнадКУЁЂЩњЮяПЩНЕНтЁЂЯИАћ№ЄИНадЁЂПЙОњадвдМАздзщзАадЃЌЪЙЕУЦфГЩЮЊНќФъРДЕФбаОПШШЕуЁЃЕЋСэвЛЗНУцЃЌЖрыФПЩФмЕФУтвпдадвВвЛжББЛИјгшИпЖШЙизЂЃЌШЛЖјжСНёУЛгаШЗЧаЕФД№АИЁЃЛљгк ЃвЃљЃсЃю злЪіжаЬсГіЕФЙлЕуПЩвдЭЦЖЯЃлЃИЃБЃнЃЌШчЙћЬхЯЕжаЕФЖрыФЗжзгСПНЯаЁЛђдкЯрЕБЖЬЪБМфФкОЭПЩБЛЩњЮяЬхЧхГ§ЃЌдђЦфЧБдкЕФУтвпдадНЯаЁЩѕжСПЩвдКіТдЃЌЯрЗДОЭашвЊПМТЧЦфЖдЩњЮяЬхЕФПЩФмУтвпдадЁЃе§ШчБОзлЪіЫљзмНсЃЌЛљгкЖрыФЕФгХСМадФмПЩЕУЕНаэЖрадФмЬиЪтЕФЫЎФ§НКЃЌдквНгУСьгђгазХЙуРЋЕФгІгУЧАОАЁЃОЁЙмКЌгаЖрыФзщЗжЕФОлКЯЮяЫЎФ§НКвбгаДѓСПЕФбаОПЃЌЕЋЮвУЧШЯЮЊетЗНУцЛЙгааэЖриНашНтОіЕФЮЪЬтЃЌБШШчЛљгкЖрыФздзщзАЕФЮяРэФ§НКСІбЇадФмВЛКУвдМАЦфЖдЭтНчЕФДЬМЄЯьгІФмСІВЛЧПЁЂОпгаЧБдкЬиЪтЙІФмЕФЖрыФиНД§ЗЂЯжЕШЕШЁЃЫфУцСйжюЖрЬєеНЃЌвВЧЁЧЁЮЊбаОПепЬсЙЉСЫИќЖрЕФбаОППеМфЃЌБШШчНЋЛЏбЇНЛСЊв§ШыЛљгкЖрыФздзщзАЕФЮяРэЫЎФ§НКжаЃЌЕЋЛЏбЇНЛСЊгжВЛПЩЙ§ЖрИЩШХЖрыФздзщзАЃЛЖрыФздзщзАаЮЪНКЭЛњРэНјааИќЩюШыбаОПЖдгкИљОнЬиЪтЪЕМЪашЧѓЩшМЦЫЎФ§НКНсЙЙОпгаживЊжИЕМвтвхЁЃЯраХЫцзХбаОПепЕФВЛаИХЌСІЃЌЛљгкЖрыФЕФОлКЯЮяЫЎФ§НКНЋЛсдкЩњЮявНгУСьгђЕУЕНИќГЄзуЕФЗЂеЙЁЃ
Утд№ЩљУїЃКБОЮФЮЊаавЕНЛСїбЇЯАЃЌАцШЈЙщдзїепМАддгжОЫљгаЃЌШчгаЧжШЈЃЌПЩСЊЯЕЩОГ§ЁЃЮФеТБъзЂгазїепМАЮФеТГіДІЃЌШчашдФЖСдЮФМАВЮПМЮФЯзЃЌПЩдФЖСддгжОЁЃ
