
摘要:实现多肽药物的口服利用,一直以来都是人们研究的热点。然而由于胃肠道复杂的生理屏障,口服多肽药物的生物利用度一直较低。本文归纳了阻碍多肽药物口服吸收的主要屏障,并综述了针对不同屏障所采取的相应治疗策略。与此同时,本文还列举了目前应用于多肽药物口服递送的新技术与新进展,这些新技术的发展将会为提高多肽药物口服生物利用度带来新希望。
相较于传统化学药物,多肽药物具有以下几点特性 :①活性较高,较少的给药剂量就能实现一定的治疗效果 ;②对酶的抵抗性低,易被胃肠道消化酶及体液中的水解酶降解,从而失去活性 ;③相对分子质量较大,且部分药物极性较强 (logP<0),较大的亲水性使其很难被细胞摄取,无法有效穿过胃肠道的生理屏障。鉴于此,多肽药物的口服生物利用度极低 [1],临床应用困难重重。目前已开发了一些技术 [ 如胃肠道渗透增强技术 (GIPET®)、瞬态渗透性增强剂 (TPE®) 技术等 ] 用于口服递送多肽药物 [2―3]。本文结合人体胃肠道的生理学特征,综述了多肽药物面对不同的生理屏障时所应用的针对性策略。
1 胃肠道的生理屏障
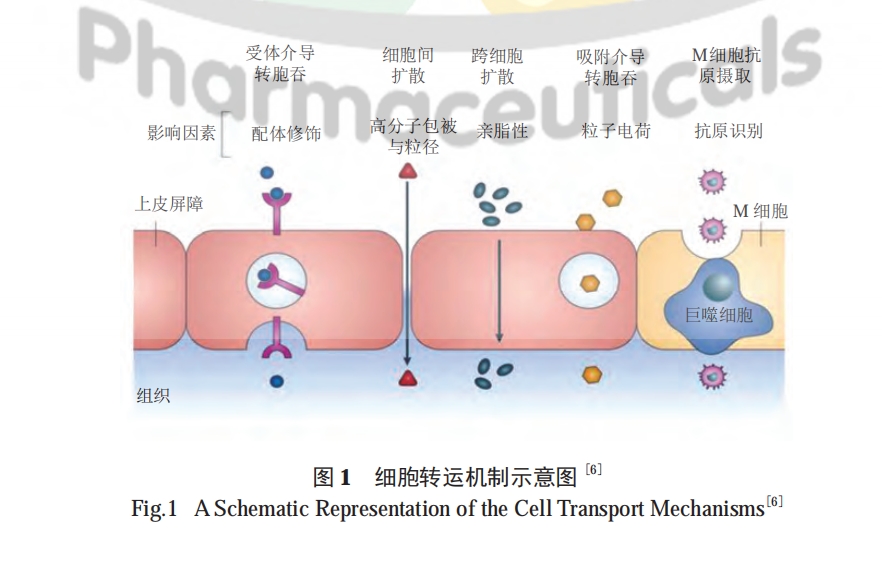
几十年来,科学家们研究了多种技术以提高多肽药物的口服生物利用度,包括使用酶抑制剂和渗透促进剂 (penetration enhancer,PE)、化学修饰、纳米载体、油相基质及结肠靶向等 [9―10]。然而,通常对于具有不同物理化学性质的多肽,需要联合应用不同的技术。下面针对不同生理屏障的应对策略进行综述。
2 针对胃部酸屏障和酶屏障的应对策略
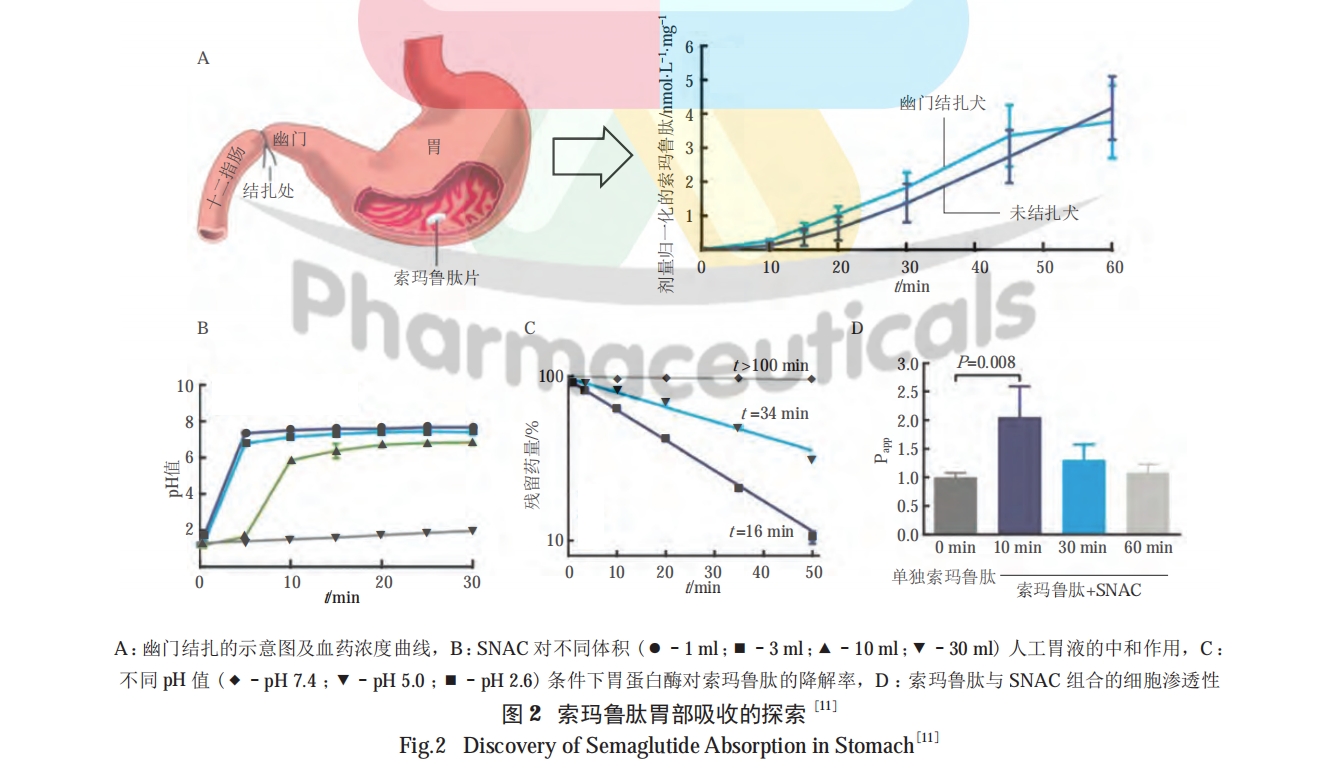
文献报道 [12],多肽与高分子连接后 [ 如聚乙二醇 (PEG) 化 ] 能有效提高稳定性,防止酶的降解,但同时也要注意高分子的加入有可能阻碍多肽与受体的结合,从而降低多肽的活性。
3 针对肠道屏障的应对策略
小肠中的酶主要是胰蛋白酶、糜蛋白酶及羧肽酶等,这些酶系能将多肽药物降解成片段,使其失去生理活性。对于这种酶屏障,最有效的手段就是加入酶抑制剂以保护多肽药物的完整结构。常用酶抑制剂包括抑肽酶、大豆蛋白酶抑制剂及亮肽素,它们能有效阻止多肽与酶的结合。酶抑制剂已被用于胰岛素的口服递送,如以色列 Oramed 公司设计的 ORMD-0801,系将蛋白酶抑制剂 ( 大豆蛋白酶抑制剂或抑肽酶 ) 与渗透促进剂 [ 乙二胺四乙酸(EDTA) 或 SNAC] 和多肽混悬在 omega-3 油酸中,进而装载于肠溶胶囊中。临床试验证明,该胶囊具有较好的口服降糖效果,已处于Ⅲ期临床试验阶段 [13]。KRAELING 等也证明抑酶肽的加入能有效提高胰岛素的生物利用度 [14]。除胰岛素外,酶抑制剂与吸收促进剂的组合还被用于降钙素及甲状旁腺激素 (PTH)(1-34) 的口服递送 [15]。然而,频繁使用酶抑制剂可能会导致过多蛋白酶的分泌,产生胰腺炎等不良反应,因此其使用的安全性有待进一步观察 [16]。
综上所述,肠道酶屏障已有较好的解决方案。除了加入酶抑制剂外,纳米载体及化学修饰等手段均能较好地保护多肽不受酶的降解。
肠上皮细胞表面附有凝胶状的黏液层,其黏度较大且在不断流动,能有效阻止外来颗粒或细菌侵入到细胞层中,有重要的保护作用,却也导致药物很难透过。黏液的不断稀释作用缩短了多肽药物与小肠上皮细胞层的接触时间,很难形成适宜药物进行渗透的浓度差。因此,研究者找到了黏膜吸附材料来实现药物的局部富集。材料与黏膜间的相互作用力主要有氢键、电荷间相互作用、疏水相互作用和范德华力。其中电荷间相互作用是一种有效且强度适中的作用力。由于黏液蛋白在肠道 pH 值条件下显电负性,因此合适的阳离子高分子聚合物能通过电荷间作用力达到黏膜黏附的目的。DAMGÉ等将聚己内酯 (PCL)与聚阳离子高分子聚合物(Eudragits RS) 混合,制备纳米粒来口服递送胰岛素,结果该纳米粒能通过电荷间相互作用紧密吸附在肠道黏膜上,最终实现 13.2%的口服生物利用度 [17]。
除了上述非共价作用力之外,硫代高分子也能与黏液中富含半胱氨酸的糖蛋白通过共价键结合产生较强的黏膜吸附作用。MARSCHÜTZ 等证明巯基化的聚丙烯酸高分子与新鲜的离体肠道黏膜的结合时间是未巯基化材料对照组的 25 倍以上 [18];KAST 等制备了巯基化比例不同的壳聚糖材料,证明巯基化程度越高,材料与黏膜的吸附能力越强 [19]。这些结果均说明富含巯基的化合物或高分子聚合物能有效地与肠道黏膜吸附,以达到富集药物的效果。
然而,仅实现黏膜吸附还无法使药物通过与小肠上皮细胞接触而被吸收,还需要使药物具有一定的黏膜渗透性 [20]。为实现黏膜渗透,就需要进行一定的修饰和改性。PEG 修饰是一种较有效的增加黏膜渗透的方法,对粒子进行 PEG 包被能有效降低纳米粒与黏膜之间的相互作用 [21]。但 PEG 的包被密度及其相对分子质量大小都会影响纳米粒的黏膜扩散能力,PEG 相对分子质量过小 (<2 000) 或过大 (>10 000) 均会影响黏膜渗透效能,且 10%PEG 包被密度的纳米粒相比于 20%及 5%时具有更好的扩散能力 [22―23]。
其他亲水性高分子材料也可增加纳米粒的黏膜内扩散,SHAN 等将胰岛素与细胞渗透肽进行混合,通过电荷间作用力形成内核,之后在表面包被一层亲水性的 N-(2- 羟丙基 ) 甲基丙烯酸共聚物 (pHPMA) 形成 170 nm 的纳米粒 [24]。由于亲水性外壳的作用,该粒子较易在黏膜中扩散,且粒子对黏膜的渗透效率也高于未包被亲水性高分子的内核。将 pHPMA 荧光标记后检测,可观察到粒子在黏膜中的荧光强度快速下降。这是因为亲水性外壳与内核通过较弱的非共价键结合,所以亲水外壳在扩散过程中就能较快脱落,在到达细胞层表面时仅剩下载药内核,不影响后续粒子的跨细胞转运。TIAN 等设计了一款具有核壳结构的纳米粒,内核由胰岛素与 N-(2- 羟丙基 )-3- 三甲基氯化铵修饰的壳聚糖构成,而外壳包被了巯基修饰的透明质酸(HA-SH)[25]。通过巯基实现粒子的黏膜吸附效果,同时随着粒子在黏液中的渗透,HA-SH 外壳不断脱落,释放出内核,继而实现对细胞的渗透。
综上所述,目前提高药物对黏液或黏膜渗透的主要方法可以概括为 :增加与黏膜的接触时间与局部药物浓度 ( 黏膜吸附 )、提高药物在黏液中的扩散 ( 黏膜渗透 ) 以及局部打开黏膜层 ( 黏液水解 )。
目前大多数的研究都集中在如何突破肠上皮细胞屏障,现有的实现细胞渗透的技术主要为多肽修饰 ( 化学改构 )、辅料添加 ( 如渗透促进剂 )、改变剂型 ( 纳米粒、微乳 ) 等,或这些技术的联合使用。
较高的 logP 值有助于药物穿透细胞膜的磷脂双分子层 [28]。因此,通过化学修饰提高多肽的亲脂性能增强其细胞渗透能力。目前,提高多肽亲脂性的化学方法为脂质化修饰,又可细分为合成前药与非前药 2 种,二者的主要区别在于是否需要通过代谢释放出活性药物,本文对此不予细分。PARMENTIER 等利用二硫键将低分子壳聚糖与艾塞那肽结合,显著提高了艾塞那肽的亲脂性及口服降糖效果 [29]。脂肪酸修饰是应用较广泛的提高多肽亲脂性的方法,其中的典型案例是口服索玛鲁肽制剂的成功开发。然而,并非所有的脂肪酸修饰都能起到增加多肽口服吸收的效果。TRIER 等分别进行了索玛鲁肽与利拉鲁肽的口服给药试验,在相同处方的情况下,口服索玛鲁肽组出现了明显的吸收效果,利拉鲁肽则几乎没有吸收,这说明并非所有的脂质化都能提高多肽的渗透性。在多肽穿透细胞的过程中存在着一种微妙的平衡,既要保证充足的与细胞膜间的相互作用,同时又不能由于膜插入程度过强而阻碍多肽的跨细胞转运 [30]。对于索玛鲁肽来说,其侧链结构为多肽与细胞膜的结合提供了合适的平衡点,换言之,有些亲脂化修饰并不一定会提高多肽渗透的能力 [11]。

综上所述,多肽的化学修饰目前主要从增加亲脂性提高被动扩散以及修饰配体增加主动转运两方面进行研究,该技术的单独应用往往并不能达到理想的体内吸收效果,需要与其他促渗方法联合应用。
降低肠上皮细胞外的钙离子浓度能扰乱胞间紧密连接中的钙离子依赖性黏着蛋白,从而打开紧密连接,增加多肽药物的渗透。钙离子螯合剂就是根据这一原理应用于促进多肽药物渗透的研究,EDTA 是其中最常用的钙离子螯合剂。Oramed 公司在艾塞那肽中加入渗透促进剂 EDTA 与大豆蛋白酶抑制剂制备肠溶制剂,实现了艾塞那肽的口服吸收,并在 Beagle 犬模型中取得了明显的口服降糖效果 [41]。一些细菌毒素能特异性地破坏紧密连接中的黏着蛋白 (E-cadherin) 及密闭蛋白 (claudine) 等,以增加多肽对上皮细胞层的渗透,如产气荚膜杆菌肠毒素 (CPE)[40]。FASANO 等将封闭小带毒素(ZOT) 应用于胰岛素口服制剂的设计,结果证明加入 ZOT 显著增强了胰岛素的渗透效率,比不加ZOT 的对照组提高了 10 倍,并且能保持小肠上皮细胞层的完整性 [42]。但打开紧密连接可能会导致一些致病微生物也跟随着多肽药物进入血液循环造成全身感染,因此具有一定的安全性风险。
综上所述,由于制备工艺简单、促渗功能较好、工艺放大容易等优点,目前渗透促进剂已得到广泛研究,相信未来将会有更多的生物大分子有望通过该技术实现口服给药。
渗透促进剂增加生物利用度的作用有限,目前较难通过该法达到 10%以上的生物利用度,因此科研人员将关注点放在了纳米载体上,希望能利用纳米载体提高多肽的吸收。纳米载体提高多肽吸收的优势在于:①纳米粒能阻止胃肠道酶对多肽的降解,起到保护多肽的作用 ;②通过调整粒径、电荷及表面性质等手段,可增加胃肠道上皮细胞对粒子的摄取 ;③对粒子表面进行主动靶向配体的修饰,能使粒子与细胞膜表面的受体或转运体特异性结合,从而完成受体介导的跨细胞膜转运,并且与直接将配体修饰在药物上的方法相比,不存在降低多肽活性的风险 ;④通过对纳米粒的制备工艺及载体材料的优化,可控制药物的释放位置及释放速率,从而有助于药物的定位、定量释放 [ 48]。基于前文所述跨上皮细胞转运的 5 个途径,纳米载体实现药物吸收主要通过以下 3 种手段 :①利用载体材料的亲脂特性,增强与细胞膜的亲和力 ;②利用各种配体 (Tf、VB12、胆酸等 )、CPP 及 Fc 片段等对载体表面进行主动靶向修饰,提高主动转运效率 ;③利用上皮细胞层上的特殊细胞 ( 如 M 细胞或杯状细胞 ) 进行主动转运。
脂类材料制成的纳米粒主要包括固体脂质纳米粒 (SLN) 及脂质体,因此利用这 2 种纳米载体进行多肽药物递送的研究较多。脂质体作为一种经典载体,也被应用到口服多肽药物递送的研究中。Diasome 公司设计了一款肝靶向的胰岛素脂质体(HDV-1),系将胰岛素包载进入粒径小于 150 nm的脂质体中,并在表面进行靶向修饰,使其能直接靶向肝脏,从而重建胰岛素在肝脏部位的正常生理响应,以达到口服降糖效果 [49]。KISEL 等开发了一款以二棕榈酰磷脂酰乙醇胺 (DPPE) 为材料的脂质体,该材料与传统蛋黄磷脂相比具有更高的熔点,因此制备的脂质体更稳定 ;体内试验证明该脂质体能实现胰岛素的有效递送,具有口服降糖效果 [50]。
除脂质体外,另一种具有代表性的脂类载体SLN 也受到广泛关注。但 SLN 只对疏水性药物具有较高的载药量,因此在包载亲水性多肽药物时就要先将药物与相应的表面活性剂通过电荷间相互作用结合,形成疏水性复合物,以提高粒子的载药量。CHEN 等利用复乳法以硬脂酸及棕榈酸甘油酯为原料制备了降钙素 SLN,药动学试验证明其口服生物利用度高达 13% [51]。CHEN 等先将艾塞那肽与胆酸钠制成胶束,再将胶束包裹在 SLN 中,这样能达到 97.7%的包封率以及 12%的口服生物利用度 [52]。
此外,还有很多天然或合成高分子材料被用于多肽药物口服递送的研究,如壳聚糖、海藻酸盐、聚乳酸 - 羟基乙酸共聚物 (PLGA)、葡聚糖等 [53]。其中壳聚糖被证明能打开紧密连接,因此应用广泛。在中性条件下,壳聚糖显正电性,能与带负电荷的高分子形成纳米粒。SONAJE 等在 pH 6.0 下将壳聚糖与聚 γ- 谷氨酸混合形成纳米粒用来负载胰岛素,冻干后装入肠溶胶囊,能达到 20%的口服生物利用度 [54]。也有人对壳聚糖纳米粒表面进行改性,将钙离子螯合剂乙二醇双 (2- 氨基乙基醚 ) 四乙酸(EGTA) 修饰到纳米粒表面,或通过在表面修饰硫醇基团以增加黏膜吸附性等手段来促进粒子经细胞间转运 [55]。总之,壳聚糖作为一种具有良好生物相容性的材料,有望在多肽药物口服递送中发挥重要作用。除上述纳米粒外,Nod 公司设计了一款具有生物黏附性的磷酸钙纳米粒,处方中包含艾塞那肽、磷酸钙及胆盐或中链脂肪酸 (C8 ~ C10),能有效增加艾塞那肽的口服吸收,目前正在进行Ⅰ期临床试验 [56]。
在纳米粒表面进行配体修饰,可促进纳米粒通过主动转运途径进行吸收,也是研究热点之一。PRIDGEN 等以聚乳酸 (PLA)-PEG 为材料制备了包载胰岛素的纳米粒,并在表面修饰了 IgG 抗体的 Fc片段,通过该片段与上皮细胞表面的 Fc 受体 (FcRn)特异性结合,介导 IgG 抗体的跨上皮细胞转运,从而实现受体介导的纳米粒摄取 [57]。FAN 等在包载胰岛素的壳聚糖 - 聚乙醇酸 (PGA) 纳米粒表面修饰去氧胆酸,通过上皮细胞表面钠离子依赖性胆酸转运体完成多肽转运 [58]。ZHANG 等制备了带有生物素靶头的脂质体,并证明该纳米粒的吸收是通过受体介导的内吞途径完成的 [59]。
在小肠细胞中,除了占多数的刷状上皮细胞外,还有一些具有特殊功能的细胞,如 M 细胞。M 细胞表面的糖蛋白较少、水解酶活性较低,因此具有较高的跨细胞膜转运能力,可转运细菌、病毒、抗原及纳米粒等 [60]。凝集素具有较好的 M 细胞靶向能力,是一类能与细胞膜上的糖脂或糖蛋白上的糖基进行特异性结合的蛋白质。在纳米粒表面进行凝集素修饰能增加 M 细胞对粒子的摄取。M 细胞表面存在的一些病原体识别受体也可被用作靶标,实现对 M 细胞的靶向作用。除靶向 M 细胞外,JIN等还报道了一种能靶向杯状细胞的壳聚糖纳米粒,该纳米粒上修饰了一段靶向肽 (CSK 肽 ),在其作用下能靶向上皮细胞层中的杯状细胞,利用特定细胞实现药物的吸收 [61]。
以上介绍的是目前常用且被广泛证明有效的一些技术。随着研究的不断深入,一些新技术也被应用于大分子药物的递送研究。离子溶液 (ionic liquids) 是一种仅由离子构成、熔点小于 100 ℃的盐。离子溶液的形成原理类似于固体的盐和共晶,是通过离子键或氢键等作用力形成的一种常温或正常体温下为液体的一种物质。此外,离子溶液具有增加难溶性药物溶解度等作用,因此也被广泛应用于小分子药物的递送 [62―63]。近年来,人们逐渐发现了离子溶液在递送生物大分子方面的潜力。BANERJEE 等利用由胆碱和香叶酸 (CAGE)组成的离子溶液制备了口服胰岛素给药体系,体外Caco-2 细胞模型试验说明 CAGE 离子溶液对胰岛素的递送具有浓度依赖性,因此推断是该制品通过打开紧密连接、增加胞间转运实现递送 [64]。同时体内试验也说明该 CAGE 离子溶液具有较好的降糖效果,且口服 5 u/kg 的胰岛素能产生与皮下注射2 u/kg 相似的吸收效果。
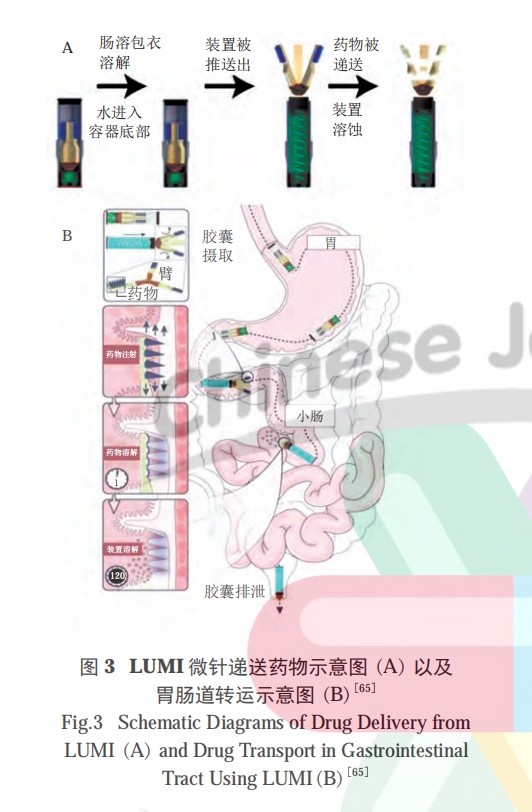
为实现胰岛素的跨膜运输,Rani 公司开发了一款基于微针技术的智能递送系统 [15]。这款智能胶囊具有一种“气球状”充气结构,当进入小肠后,外层肠溶衣壳开始溶解,触发枸橼酸和碳酸氢钠的反应,释放出二氧化碳,使胶囊内部结构膨胀,将微针刺入到小肠肠腔上,从而实现胰岛素的跨小肠上皮细胞释放。这种智能胶囊直接穿透了肠壁的物理屏障,因此递送效率也得到较大提高,生物利用度可达到 50%以上,成为一种递送多肽药物的新式“武器”。
4 结论

多肽药物口服递送面临着众多的生物屏障,无论是酸屏障、酶屏障,还是肠壁细胞层屏障,目前都已找到了较好的解决手段。然而目前上市的口服多肽产品有限,大部分研究仍然处在临床前或临床试验阶段。酶抑制剂与渗透促进剂的组合在临床试验中被证明是一种有效的递送手段,但该法对于生物利用度的提高并不十分理想。较低的生物利用度往往会浪费较多药物,导致成本提高。而纳米粒虽能达到较高的生物利用度,但面临着产业化难度大、批间差异显著等缺陷,较难实现临床应用。一些新技术 ( 离子溶液、微针 ) 的应用让人们看到了多肽药物口服给药的新希望。
由于多肽的物理化学性质千差万别,每一种多肽药物都应有其最适合的递送方式。SNAC 能有效递送索玛鲁肽但却无法实现利拉鲁肽的口服吸收这一实例提醒我们,针对不同性质的药物要选择不同的口服递送策略 [11]。现有的策略均有其一定的局限性,或是无法实现生物利用度的更大提升,或是无法在工艺水平上实现量产。
随着人们对多肽药物在胃肠道生理环境下行为的深入研究及递送药物技术的不断进步,在多肽药物口服递送领域中的尝试会越来越多,相信将来会有更多能提高患者顺应性的口服多肽产品问世,为慢性病患者带来福音。
免责声明:本文为行业交流学习,版权归原作者及原杂志所有,如有侵权,可联系删除。文章标注有作者及文章出处,如需阅读原文及参考文献,可阅读原杂志。

电话:0551-65177703 邮箱:pb@peptidesbank.com 地址:安徽省合肥市四川路868号云谷创新园A6栋3层
合肥肽库生物(Taikubio)只为有资质的科研机构、医药企业基于科学研究或药证申报的用途提供医药研发服务, 不为任何个人或者非科研性质的、非用于药证申报使用等其他用途提供服务。