
’Σ“ΣΘΚ Βœ÷ΕύκΡ“©ΈοΒΡΩΎΖΰάϊ”ΟΘ§“Μ÷±“‘ά¥ΕΦ «»ΥΟ«―–ΨΩΒΡ»»ΒψΓΘ»ΜΕχ”…”ΎΈΗ≥ΠΒάΗ¥‘”ΒΡ…ζάμΤΝ’œΘ§ΩΎΖΰΕύκΡ“©ΈοΒΡ…ζΈοάϊ”ΟΕ»“Μ÷±ΫœΒΆΓΘ±ΨΈΡΙιΡ…ΝΥΉηΑ≠ΕύκΡ“©ΈοΩΎΖΰΈϋ ’ΒΡ÷ς“ΣΤΝ’œΘ§≤ΔΉέ ωΝΥ’κΕ‘≤ΜΆ§ΤΝ’œΥυ≤…»ΓΒΡœύ”Π÷ΈΝΤ≤Ώ¬‘ΓΘ”κ¥ΥΆ§ ±Θ§±ΨΈΡΜΙΝ–ΨΌΝΥΡΩ«Α”Π”Ο”ΎΕύκΡ“©ΈοΩΎΖΰΒίΥΆΒΡ–¬ΦΦ θ”κ–¬Ϋχ’ΙΘ§’β–©–¬ΦΦ θΒΡΖΔ’ΙΫΪΜαΈΣΧαΗΏΕύκΡ“©ΈοΩΎΖΰ…ζΈοάϊ”ΟΕ»¥χά¥–¬œΘΆϊΓΘ
œύΫœ”Ύ¥ΪΆ≥Μ·―ß“©ΈοΘ§ΕύκΡ“©ΈοΨΏ”–“‘œ¬ΦΗΒψΧΊ–‘ ΘΚΔΌΜν–‘ΫœΗΏΘ§Ϋœ…ΌΒΡΗχ“©ΦΝΝΩΨΆΡή Βœ÷“ΜΕ®ΒΡ÷ΈΝΤ–ßΙϊ ΘΜΔΎΕ‘ΟΗΒΡΒ÷ΩΙ–‘ΒΆΘ§“Ή±ΜΈΗ≥ΠΒάœϊΜ·ΟΗΦΑΧε“Κ÷–ΒΡΥ°ΫβΟΗΫΒΫβΘ§¥”Εχ ß»ΞΜν–‘ ΘΜΔέœύΕ‘Ζ÷Ή”÷ ΝΩΫœ¥σΘ§«“≤ΩΖ÷“©ΈοΦΪ–‘Ϋœ«Ω (logP<0)Θ§Ϋœ¥σΒΡ«ΉΥ°–‘ ΙΤδΚήΡ―±ΜœΗΑϊ…ψ»ΓΘ§ΈόΖ®”––ߥ©ΙΐΈΗ≥ΠΒάΒΡ…ζάμΤΝ’œΓΘΦχ”Ύ¥ΥΘ§ΕύκΡ“©ΈοΒΡΩΎΖΰ…ζΈοάϊ”ΟΕ»ΦΪΒΆ [1]Θ§ΝΌ¥≤”Π”ΟάßΡ―÷Ί÷ΊΓΘΡΩ«Α“―ΩΣΖΔΝΥ“Μ–©ΦΦ θ [ »γΈΗ≥ΠΒά…χΆΗ‘ω«ΩΦΦ θ (GIPET®)ΓΔΥ≤Χ§…χΆΗ–‘‘ω«ΩΦΝ (TPE®) ΦΦ θΒ» ] ”Ο”ΎΩΎΖΰΒίΥΆΕύκΡ“©Έο [2ΓΣ3]ΓΘ±ΨΈΡΫαΚœ»ΥΧεΈΗ≥ΠΒάΒΡ…ζάμ―ßΧΊ’ςΘ§Ήέ ωΝΥΕύκΡ“©ΈοΟφΕ‘≤ΜΆ§ΒΡ…ζάμΤΝ’œ ±Υυ”Π”ΟΒΡ’κΕ‘–‘≤Ώ¬‘ΓΘ
1 ΈΗ≥ΠΒάΒΡ…ζάμΤΝ’œ
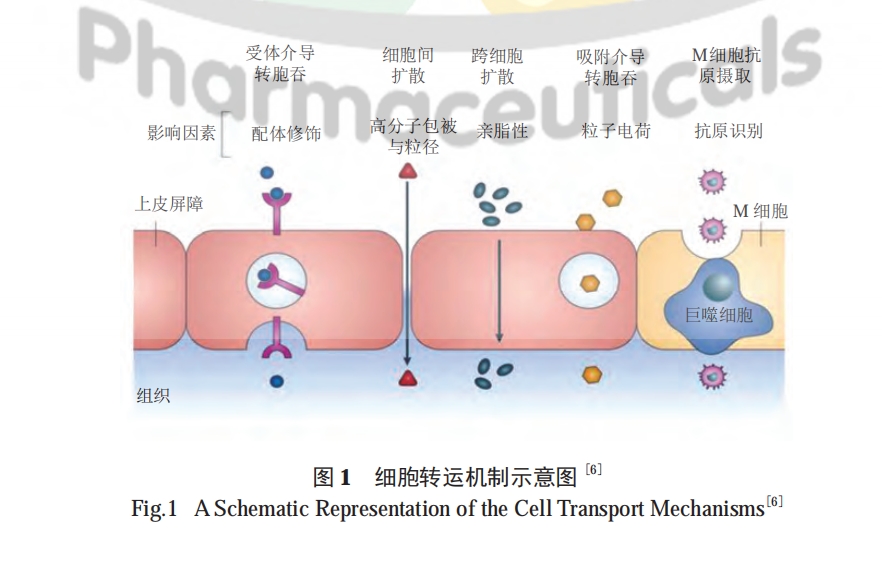
ΑιΥφΉ≈“©ΈοΆ®ΙΐΩΎ«ΜΫχ»κΒΫ»ΥΒΡΈΗ≥ΠΒάœΒΆ≥Θ§ΕύκΡ“©ΈοΥυΟφΝΌΒΡΒΎ“ΜΒάΤΝ’œΨΆ «ΈΗ≤ΩΒΡΥα”κΟΗΜΖΨ≥ΓΘΈΗ≤ΩΒΡΦΪΕΥΥα–‘ΜΖΨ≥Μα ΙΕύκΡΖΔ…ζ÷ Ή”Μ·Μρ¥ρΩΣΕύκΡΒΡ’έΒΰΘ§ ΙΕύκΡ ß»ΞΩ’ΦδΫαΙΙΘ§ΫχΕχ…Ξ ß…ζάμΜν–‘ [4]Θ§Ά§ ±ΈΗΒΑΑΉΟΗ‘ΎΥα–‘ΜΖΨ≥÷–Ρή ΙΕύκΡΖΔ…ζΟΗΫβΓΘ÷°ΚσΘ§ΕύκΡ“©ΈοΫΪΫχ»κΒΫ“©ΈοΈϋ ’ΒΡ÷Ί“Σ≤ΩΈΜΓΣΓΣ–Γ≥ΠΓΘ–Γ≥Π≤ΩΈΜ pH ÷ΒΤΪ÷––‘Θ§ΨΏ”–±»ΈΗΗϋΗ¥‘”ΒΡΟΗΜΖΨ≥ ( »γΟ”ΒΑΑΉΟΗΓΔτ»κΡΟΗΒ» )Θ§Ρή ΙΕύκΡΟΗΫβ≥…–ΓΤ§ΕΈΕχ ßΜν [ 5]ΓΘΦ¥±ψΧ”Ά―ΝΥΟΗΫβΘ§ΕύκΡΈϋ ’ΜΙ–η“ΣΟφΕ‘Ήν÷ς“Σ“≤ «ΉνΡ―ΩΥΖΰΒΡ“ΜΒάΤΝ’œΓΣΓΣΫτΟήΒΡ–Γ≥Π…œΤΛœΗΑϊ≤ψΓΘΫτΟήΝ§Ϋ”ΒΡ–Γ≥Π…œΤΛœΗΑϊ…œΗΫΉ≈”–ΡΐΫΚΉ¥ΒΡΓΔ÷ς“Σ”…Χ«ΒΑΑΉΙΙ≥…ΒΡπΛ“Κ≤ψΘ§”…”ΎΤδπΛΕ»ΫœΗΏΘ§«“Ρή”κΕύκΡ≤ζ…ζ«βΦϋΓΔΖΕΒ¬ΜΣΝΠΜρΒγΚ…ΦδœύΜΞΉς”ΟΘ§¥”Εχ ΙΕύκΡΒΡά©…Δ±δΒΟ °Ζ÷άßΡ―ΓΘ‘ΎπΛΡΛ≤ψœ¬ΨΆ «ΈΗ≥ΠΒάΒΡΉνΚσ“ΜΒάΤΝ’œΓΣΓΣ≥ΠΒά…œΤΛœΗΑϊ≤ψΓΘΡΩ«Α―–ΨΩ±μΟςΘ§“©Έο¥©Ιΐ≥ΠΒά…œΤΛœΗΑϊ≤ψ÷ς“Σ”–“‘œ¬ 5 ΗωΆΨΨΕ ( ΆΦ 1[6]) ΘΚΔΌΩγœΗΑϊ±ΜΕ·ΉΣ‘ΥΆΨΨΕΘ§¥σ≤ΩΖ÷«Ή÷§–‘–ΓΖ÷Ή”“©ΈοΕΦΆ®Ιΐ’β“ΜΆΨΨΕ Βœ÷ΩγœΗΑϊΉΣ‘Υ ΘΜΔΎ ήΧεΫιΒΦΒΡΉΣΑϊΆΧΆΨΨΕΘ§–ό Έ”–≈δΧεΒΡ“©ΈοΡή”κ…œΤΛœΗΑϊ±μ¥οΒΡ ήΧεΖΔ…ζΧΊ“λ–‘ΫαΚœΘ§ Βœ÷ ήΧεΫιΒΦΒΡΉΣΑϊΆΧΉς”Ο ΘΜΔέΈϋΗΫΫιΒΦΒΡΉΣΑϊΆΧΆΨΨΕΘ§”…”ΎœΗΑϊΡΛ±μΟφ≥ ΒγΗΚ–‘Θ§“ρ¥Υ―τάκΉ”“©ΈοΜρΡ…ΟΉΝΘΡήΆ®ΙΐΈϋΗΫΫιΒΦΉΣΑϊΆΧΒΡΖΫ ΫΆξ≥…ΉΣ‘Υ ΘΜΔή≈‘¬ΖΉΣ‘ΥΘ§”…”Ύ…œΤΛœΗΑϊ÷°Φδ¥φ‘ΎΉ≈ΫτΟήΝ§Ϋ”Θ§œΗΑϊΦδΒΡΦδœΕΚή–Γ (100 nm)Θ§“ρ¥Υ÷Μ”–“Μ–©«ΉΥ°–‘–ΓΖ÷Ή”“©Έο≤≈ΡήΆ®Ιΐ ΘΜΔί MœΗΑϊΉΣ‘ΥΘ§‘Ύ–Γ≥Π…œΤΛœΗΑϊ÷–¥φ‘ΎΉ≈“ΜάύΧΊ βΒΡœΗΑϊΓΣΓΣM œΗΑϊΘ§÷ς“ΣΙΠΡή «…ψ»Γ≤ΔΉΣ‘ΥΩΙ‘≠Θ§ ΙΤδΫχ»κΝήΑΆ―≠ΜΖΘ§≤Έ”κΒΫ»Ϊ…μ―≠ΜΖ÷– [7]ΓΘ”…”ΎΕύκΡΆ®≥ΘΨΏ”–Ϋœ¥σΒΡœύΕ‘Ζ÷Ή”÷ ΝΩΓΔΫœ«ΩΒΡ«ΉΥ°–‘Θ§ΤδΩγΡΛΉΣ‘Υ °Ζ÷άßΡ― [8]ΓΘ
ΦΗ °Ρξά¥Θ§ΩΤ―ßΦ“Ο«―–ΨΩΝΥΕύ÷÷ΦΦ θ“‘ΧαΗΏΕύκΡ“©ΈοΒΡΩΎΖΰ…ζΈοάϊ”ΟΕ»Θ§Αϋά® Ι”ΟΟΗ“÷÷ΤΦΝΚΆ…χΆΗ¥ΌΫχΦΝ (penetration enhancerΘ§PE)ΓΔΜ·―ß–ό ΈΓΔΡ…ΟΉ‘ΊΧεΓΔ”ΆœύΜυ÷ ΦΑΫα≥ΠΑ–œρΒ» [9ΓΣ10]ΓΘ»ΜΕχΘ§Ά®≥ΘΕ‘”ΎΨΏ”–≤ΜΆ§ΈοάμΜ·―ß–‘÷ ΒΡΕύκΡΘ§–η“ΣΝΣΚœ”Π”Ο≤ΜΆ§ΒΡΦΦ θΓΘœ¬Οφ’κΕ‘≤ΜΆ§…ζάμΤΝ’œΒΡ”ΠΕ‘≤Ώ¬‘Ϋχ––Ήέ ωΓΘ
2 ’κΕ‘ΈΗ≤ΩΥαΤΝ’œΚΆΟΗΤΝ’œΒΡ”ΠΕ‘≤Ώ¬‘
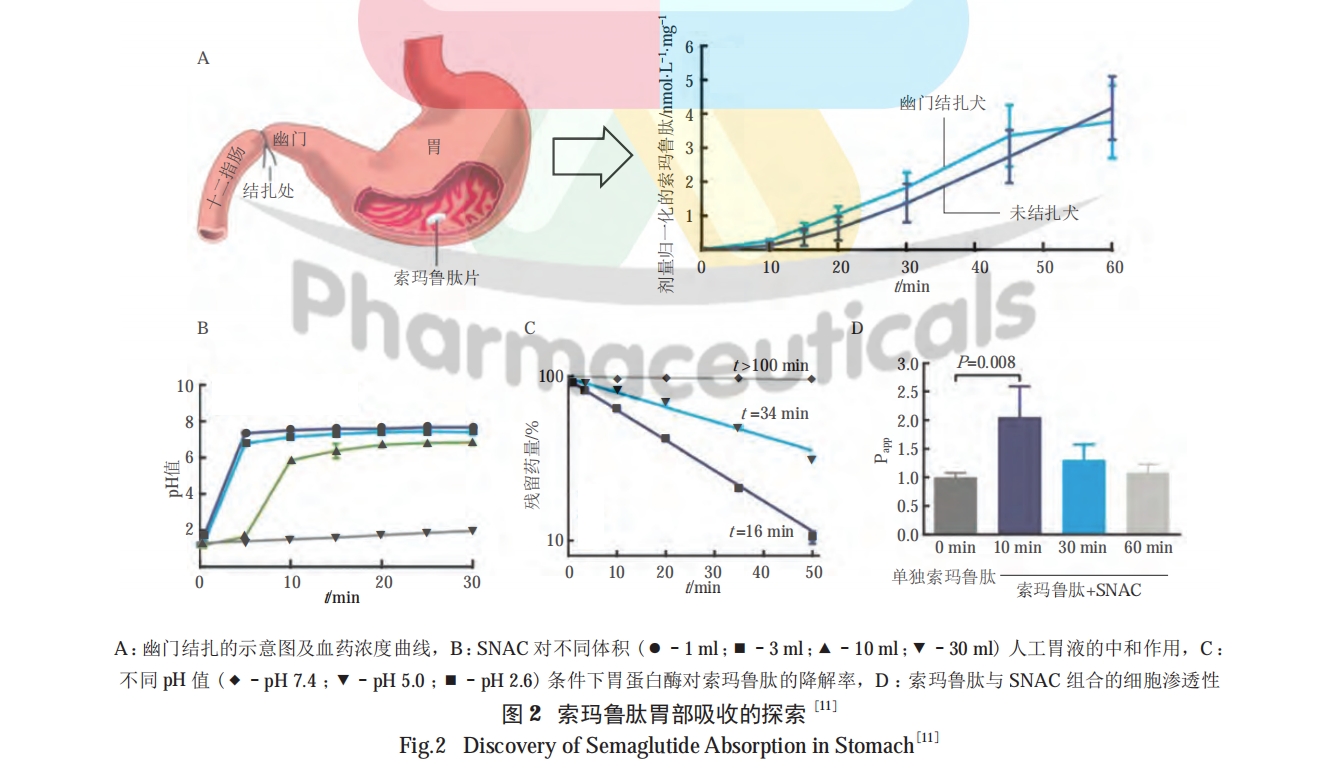
Ά®≥Θ÷Τ±ΗΩΎΖΰΕύκΡ“©Έο÷ΤΦΝ ±ΆυΆυάϊ”Ο≥Π»ήΑϋ“¬Μρ≥Π»ήΫΚΡ“Β» ÷ΕΈΘ§±ήΟβΤδ‘ΎΈΗ≤Ω ΆΖ≈ ±±ΜΫΒΫβΓΘ»ΜΕχΘ§»ΥΟ«Κω¬‘ΝΥΈΗΕ‘“©ΈοΒΡΈϋ ’ΡήΝΠΘ§«“ΈΗ≤ΩΈϋ ’ ±ΜΙΨΏ”–“©Έο”κΈΗΒΡΫ”¥Ξ ±ΦδΫœΕΧΓΔΟΗΜΖΨ≥œύΕ‘ΦρΒΞΒΡΧΊΒψΓΘ“ρ¥ΥΘ§÷Μ“ΣΫβΨωΥα”κΟΗΝΫ¥σΤΝ’œΘ§ΨΆΩ…Ρή Βœ÷ΕύκΡ‘ΎΈΗ≤ΩΒΡΈϋ ’ΓΘ2019 ΡξΟάΙζ FDA ≈ζΉΦ…œ –ΒΡΩΎΖΰΥς¬ξ¬≥κΡΤ§ ( ”Ο”Ύ÷ΈΝΤ 2 –ΆΧ«Ρρ≤Γ )ΨΆ±Μ÷Λ Β «Ω…“‘‘ΎΈΗ≤ΩΆξ≥…Έϋ ’Ιΐ≥ΧΒΡ [11]ΓΘ‘Ύ“‘Beagle »°ΈΣΡΘ–ΆΒΡΕ·Έο ‘―ι÷–Θ§“‘―ΣΫ§÷–“©Έο≈®Ε»ΈΣΤάΦέ÷Η±ξΘ§Ε‘±»ΝΥ”ΡΟ≈Ϋα‘ζΚΆ≤ΜΫα‘ζΒΡ«ιΩωœ¬Θ§Τ§ΦΝ [ Φ¥Υς¬ξ¬≥κΡ”κ…χΆΗ¥ΌΫχΦΝ 8-(2- τ«Μυ±ΫΦΉθΘΑΖΜυ ) –ΝΥαΡΤ (SNAC) Ι≤Μλ÷Τ≥…ΒΡΤ’Ά®Τ§ ] ÷–ΒΡ“©Έο‘ΎΈΗ≤ΩΒΡΈϋ ’«ιΩωΓΘΫαΙϊœ‘ Ψ 2 ÷÷«ιΩωœ¬―Σ“©≈®Ε»ΈόΟςœ‘≤ν“λΘ§ΥΒΟςΥς¬ξ¬≥κΡΫωΩΩΈΗ≤ΩΈϋ ’ΨΆΡή≤ζ…ζ”κ»ΪΈΗ≥ΠΒάΈϋ ’œύΥΤΒΡ–ßΙϊΘ§÷ΛΟςΝΥΈΗ «Υς¬ξ¬≥κΡΒΡΈϋ ’≤ΩΈΜΒΡΫα¬έΓΘΆ§ ±Θ§Ής’ΏΧΫΥςΝΥΗΟΩΎΖΰ÷ΤΦΝΙφ±ήΈΗ÷–ΟΗ”κΥαΥΪ÷ΊΫΒΫβΒΡΜζ÷ΤΓΘΫΪΤ§ΦΝΆΕ»κΒΫ»ΥΙΛΈΗ“Κ ( Κ§ΈΗΒΑΑΉΟΗ ) ÷–Θ§”…”Ύ SNAC ΨΏ”–“ΜΕ®ΒΡΜΚ≥εΡήΝΠΘ§Ρή÷±Ϋ”ΫΪΫι÷ pH ÷ΒΧαΗΏΒΫ÷––‘Θ§ΕχΈΗΒΑΑΉΟΗ‘Ύ÷––‘Ϋι÷ ÷–Μα ßΜνΘ§“ρ¥ΥΥς¬ξ¬≥κΡΨΆ±ήΟβΝΥ±ΜΈΗΥα“‘ΦΑΈΗΒΑΑΉΟΗΫΒΫβΘ§¥”ΕχΈΣΕύκΡ‘ΎΈΗ≤ΩΒΡΈϋ ’ΧαΙ©ΝΥΩ…Ρή ( »γΆΦ 2)ΓΘ
ΈΡœΉ±®Βά [12]Θ§ΕύκΡ”κΗΏΖ÷Ή”Ν§Ϋ”Κσ [ »γΨέ““Εΰ¥Φ (PEG) Μ· ] Ρή”––ßΧαΗΏΈ»Ε®–‘Θ§Ζά÷ΙΟΗΒΡΫΒΫβΘ§ΒΪΆ§ ±“≤“ΣΉΔ“βΗΏΖ÷Ή”ΒΡΦ”»κ”–Ω…ΡήΉηΑ≠ΕύκΡ”κ ήΧεΒΡΫαΚœΘ§¥”ΕχΫΒΒΆΕύκΡΒΡΜν–‘ΓΘ
3 ’κΕ‘≥ΠΒάΤΝ’œΒΡ”ΠΕ‘≤Ώ¬‘
3.1 ≥ΠΒάΟΗΤΝ’œ
–Γ≥Π÷–ΒΡΟΗ÷ς“Σ «“»ΒΑΑΉΟΗΓΔΟ”ΒΑΑΉΟΗΦΑτ»κΡΟΗΒ»Θ§’β–©ΟΗœΒΡήΫΪΕύκΡ“©ΈοΫΒΫβ≥…Τ§ΕΈΘ§ ΙΤδ ß»Ξ…ζάμΜν–‘ΓΘΕ‘”Ύ’β÷÷ΟΗΤΝ’œΘ§Ήν”––ßΒΡ ÷ΕΈΨΆ «Φ”»κΟΗ“÷÷ΤΦΝ“‘±ΘΜΛΕύκΡ“©ΈοΒΡΆξ’ϊΫαΙΙΓΘ≥Θ”ΟΟΗ“÷÷ΤΦΝΑϋά®“÷κΡΟΗΓΔ¥σΕΙΒΑΑΉΟΗ“÷÷ΤΦΝΦΑΝΝκΡΥΊΘ§ΥϋΟ«Ρή”––ßΉη÷ΙΕύκΡ”κΟΗΒΡΫαΚœΓΘΟΗ“÷÷ΤΦΝ“―±Μ”Ο”Ύ“»ΒΚΥΊΒΡΩΎΖΰΒίΥΆΘ§»γ“‘…ΪΝ– Oramed ΙΪΥΨ…ηΦΤΒΡ ORMD-0801Θ§œΒΫΪΒΑΑΉΟΗ“÷÷ΤΦΝ ( ¥σΕΙΒΑΑΉΟΗ“÷÷ΤΦΝΜρ“÷κΡΟΗ ) ”κ…χΆΗ¥ΌΫχΦΝ [ ““ΕΰΑΖΥΡ““Υα(EDTA) Μρ SNAC] ΚΆΕύκΡΜλ–ϋ‘Ύ omega-3 ”ΆΥα÷–Θ§ΫχΕχΉΑ‘Ί”Ύ≥Π»ήΫΚΡ“÷–ΓΘΝΌ¥≤ ‘―ι÷ΛΟςΘ§ΗΟΫΚΡ“ΨΏ”–ΫœΚΟΒΡΩΎΖΰΫΒΧ«–ßΙϊΘ§“―¥Π”ΎΔσΤΎΝΌ¥≤ ‘―ιΫΉΕΈ [13]ΓΘKRAELING Β»“≤÷ΛΟς“÷ΟΗκΡΒΡΦ”»κΡή”––ßΧαΗΏ“»ΒΚΥΊΒΡ…ζΈοάϊ”ΟΕ» [14]ΓΘ≥ΐ“»ΒΚΥΊΆβΘ§ΟΗ“÷÷ΤΦΝ”κΈϋ ’¥ΌΫχΦΝΒΡΉιΚœΜΙ±Μ”Ο”ΎΫΒΗΤΥΊΦΑΦΉΉ¥≈‘œΌΦΛΥΊ (PTH)(1-34) ΒΡΩΎΖΰΒίΥΆ [15]ΓΘ»ΜΕχΘ§ΤΒΖ± Ι”ΟΟΗ“÷÷ΤΦΝΩ…ΡήΜαΒΦ÷¬ΙΐΕύΒΑΑΉΟΗΒΡΖ÷ΟΎΘ§≤ζ…ζ“»œΌ―ΉΒ»≤ΜΝΦΖ¥”ΠΘ§“ρ¥ΥΤδ Ι”ΟΒΡΑ≤»Ϊ–‘”–¥ΐΫχ“Μ≤ΫΙέ≤λ [16]ΓΘ
Ήέ…œΥυ ωΘ§≥ΠΒάΟΗΤΝ’œ“―”–ΫœΚΟΒΡΫβΨωΖΫΑΗΓΘ≥ΐΝΥΦ”»κΟΗ“÷÷ΤΦΝΆβΘ§Ρ…ΟΉ‘ΊΧεΦΑΜ·―ß–ό ΈΒ» ÷ΕΈΨυΡήΫœΚΟΒΊ±ΘΜΛΕύκΡ≤Μ ήΟΗΒΡΫΒΫβΓΘ
3.2 πΛ“ΚΜρπΛΡΛΤΝ’œ
≥Π…œΤΛœΗΑϊ±μΟφΗΫ”–ΡΐΫΚΉ¥ΒΡπΛ“Κ≤ψΘ§ΤδπΛΕ»Ϋœ¥σ«“‘Ύ≤ΜΕœΝςΕ·Θ§Ρή”––ßΉη÷ΙΆβά¥Ω≈ΝΘΜρœΗΨζ«÷»κΒΫœΗΑϊ≤ψ÷–Θ§”–÷Ί“ΣΒΡ±ΘΜΛΉς”ΟΘ§»¥“≤ΒΦ÷¬“©ΈοΚήΡ―ΆΗΙΐΓΘπΛ“ΚΒΡ≤ΜΕœœΓ ΆΉς”ΟΥθΕΧΝΥΕύκΡ“©Έο”κ–Γ≥Π…œΤΛœΗΑϊ≤ψΒΡΫ”¥Ξ ±ΦδΘ§ΚήΡ―–Έ≥… “Υ“©ΈοΫχ––…χΆΗΒΡ≈®Ε»≤νΓΘ“ρ¥ΥΘ§―–ΨΩ’Ώ’“ΒΫΝΥπΛΡΛΈϋΗΫ≤ΡΝœά¥ Βœ÷“©ΈοΒΡΨ÷≤ΩΗΜΦ·ΓΘ≤ΡΝœ”κπΛΡΛΦδΒΡœύΜΞΉς”ΟΝΠ÷ς“Σ”–«βΦϋΓΔΒγΚ…ΦδœύΜΞΉς”ΟΓΔ ηΥ°œύΜΞΉς”ΟΚΆΖΕΒ¬ΜΣΝΠΓΘΤδ÷–ΒγΚ…ΦδœύΜΞΉς”Ο «“Μ÷÷”––ß«“«ΩΕ» ÷–ΒΡΉς”ΟΝΠΓΘ”…”ΎπΛ“ΚΒΑΑΉ‘Ύ≥ΠΒά pH ÷ΒΧθΦΰœ¬œ‘ΒγΗΚ–‘Θ§“ρ¥ΥΚœ ΒΡ―τάκΉ”ΗΏΖ÷Ή”ΨέΚœΈοΡήΆ®ΙΐΒγΚ…ΦδΉς”ΟΝΠ¥οΒΫπΛΡΛπΛΗΫΒΡΡΩΒΡΓΘDAMGÉΒ»ΫΪΨέΦΚΡΎθΞ (PCL)”κΨέ―τάκΉ”ΗΏΖ÷Ή”ΨέΚœΈο(Eudragits RS) ΜλΚœΘ§÷Τ±ΗΡ…ΟΉΝΘά¥ΩΎΖΰΒίΥΆ“»ΒΚΥΊΘ§ΫαΙϊΗΟΡ…ΟΉΝΘΡήΆ®ΙΐΒγΚ…ΦδœύΜΞΉς”ΟΫτΟήΈϋΗΫ‘Ύ≥ΠΒάπΛΡΛ…œΘ§Ήν÷’ Βœ÷ 13.2ΘΞΒΡΩΎΖΰ…ζΈοάϊ”ΟΕ» [17]ΓΘ
≥ΐΝΥ…œ ωΖ«Ι≤ΦέΉς”ΟΝΠ÷°ΆβΘ§Νρ¥ζΗΏΖ÷Ή”“≤Ρή”κπΛ“Κ÷–ΗΜΚ§ΑκκΉΑ±ΥαΒΡΧ«ΒΑΑΉΆ®ΙΐΙ≤ΦέΦϋΫαΚœ≤ζ…ζΫœ«ΩΒΡπΛΡΛΈϋΗΫΉς”ΟΓΘMARSCHÜTZ Β»÷ΛΟςέœΜυΜ·ΒΡΨέ±ϊœ©ΥαΗΏΖ÷Ή””κ–¬œ ΒΡάκΧε≥ΠΒάπΛΡΛΒΡΫαΚœ ±Φδ «Έ¥έœΜυΜ·≤ΡΝœΕ‘’’ΉιΒΡ 25 ±Ε“‘…œ [18]ΘΜKAST Β»÷Τ±ΗΝΥέœΜυΜ·±»άΐ≤ΜΆ§ΒΡΩ«ΨέΧ«≤ΡΝœΘ§÷ΛΟςέœΜυΜ·≥ΧΕ»‘ΫΗΏΘ§≤ΡΝœ”κπΛΡΛΒΡΈϋΗΫΡήΝΠ‘Ϋ«Ω [19]ΓΘ’β–©ΫαΙϊΨυΥΒΟςΗΜΚ§έœΜυΒΡΜ·ΚœΈοΜρΗΏΖ÷Ή”ΨέΚœΈοΡή”––ßΒΊ”κ≥ΠΒάπΛΡΛΈϋΗΫΘ§“‘¥οΒΫΗΜΦ·“©ΈοΒΡ–ßΙϊΓΘ
»ΜΕχΘ§Ϋω Βœ÷πΛΡΛΈϋΗΫΜΙΈόΖ® Ι“©ΈοΆ®Ιΐ”κ–Γ≥Π…œΤΛœΗΑϊΫ”¥ΞΕχ±ΜΈϋ ’Θ§ΜΙ–η“Σ Ι“©ΈοΨΏ”–“ΜΕ®ΒΡπΛΡΛ…χΆΗ–‘ [20]ΓΘΈΣ Βœ÷πΛΡΛ…χΆΗΘ§ΨΆ–η“ΣΫχ––“ΜΕ®ΒΡ–ό ΈΚΆΗΡ–‘ΓΘPEG –ό Έ «“Μ÷÷Ϋœ”––ßΒΡ‘ωΦ”πΛΡΛ…χΆΗΒΡΖΫΖ®Θ§Ε‘ΝΘΉ”Ϋχ–– PEG Αϋ±ΜΡή”––ßΫΒΒΆΡ…ΟΉΝΘ”κπΛΡΛ÷°ΦδΒΡœύΜΞΉς”Ο [21]ΓΘΒΪ PEG ΒΡΑϋ±ΜΟήΕ»ΦΑΤδœύΕ‘Ζ÷Ή”÷ ΝΩ¥σ–ΓΕΦΜα”ΑœλΡ…ΟΉΝΘΒΡπΛΡΛά©…ΔΡήΝΠΘ§PEG œύΕ‘Ζ÷Ή”÷ ΝΩΙΐ–Γ (<2 000) ΜρΙΐ¥σ (>10 000) ΨυΜα”ΑœλπΛΡΛ…χΆΗ–ßΡήΘ§«“ 10ΘΞPEG Αϋ±ΜΟήΕ»ΒΡΡ…ΟΉΝΘœύ±»”Ύ 20ΘΞΦΑ 5ΘΞ ±ΨΏ”–ΗϋΚΟΒΡά©…ΔΡήΝΠ [22ΓΣ23]ΓΘ
ΤδΥϊ«ΉΥ°–‘ΗΏΖ÷Ή”≤ΡΝœ“≤Ω…‘ωΦ”Ρ…ΟΉΝΘΒΡπΛΡΛΡΎά©…ΔΘ§SHAN Β»ΫΪ“»ΒΚΥΊ”κœΗΑϊ…χΆΗκΡΫχ––ΜλΚœΘ§Ά®ΙΐΒγΚ…ΦδΉς”ΟΝΠ–Έ≥…ΡΎΚΥΘ§÷°Κσ‘Ύ±μΟφΑϋ±Μ“Μ≤ψ«ΉΥ°–‘ΒΡ N-(2- τ«±ϊΜυ ) ΦΉΜυ±ϊœ©ΥαΙ≤ΨέΈο (pHPMA) –Έ≥… 170 nm ΒΡΡ…ΟΉΝΘ [24]ΓΘ”…”Ύ«ΉΥ°–‘ΆβΩ«ΒΡΉς”ΟΘ§ΗΟΝΘΉ”Ϋœ“Ή‘ΎπΛΡΛ÷–ά©…ΔΘ§«“ΝΘΉ”Ε‘πΛΡΛΒΡ…χΆΗ–߬ “≤ΗΏ”ΎΈ¥Αϋ±Μ«ΉΥ°–‘ΗΏΖ÷Ή”ΒΡΡΎΚΥΓΘΫΪ pHPMA ”ΪΙβ±ξΦ«ΚσΦλ≤βΘ§Ω…Ιέ≤λΒΫΝΘΉ”‘ΎπΛΡΛ÷–ΒΡ”ΪΙβ«ΩΕ»ΩλΥΌœ¬ΫΒΓΘ’β «“ρΈΣ«ΉΥ°–‘ΆβΩ«”κΡΎΚΥΆ®ΙΐΫœ»θΒΡΖ«Ι≤ΦέΦϋΫαΚœΘ§Υυ“‘«ΉΥ°ΆβΩ«‘Ύά©…ΔΙΐ≥Χ÷–ΨΆΡήΫœΩλΆ―¬δΘ§‘ΎΒΫ¥οœΗΑϊ≤ψ±μΟφ ±Ϋω Θœ¬‘Ί“©ΡΎΚΥΘ§≤Μ”ΑœλΚσ–χΝΘΉ”ΒΡΩγœΗΑϊΉΣ‘ΥΓΘTIAN Β»…ηΦΤΝΥ“ΜΩνΨΏ”–ΚΥΩ«ΫαΙΙΒΡΡ…ΟΉΝΘΘ§ΡΎΚΥ”…“»ΒΚΥΊ”κ N-(2- τ«±ϊΜυ )-3- »ΐΦΉΜυ¬»Μ·οß–ό ΈΒΡΩ«ΨέΧ«ΙΙ≥…Θ§ΕχΆβΩ«Αϋ±ΜΝΥέœΜυ–ό ΈΒΡΆΗΟς÷ Υα(HA-SH)[25]ΓΘΆ®ΙΐέœΜυ Βœ÷ΝΘΉ”ΒΡπΛΡΛΈϋΗΫ–ßΙϊΘ§Ά§ ±ΥφΉ≈ΝΘΉ”‘ΎπΛ“Κ÷–ΒΡ…χΆΗΘ§HA-SH ΆβΩ«≤ΜΕœΆ―¬δΘ§ ΆΖ≈≥ωΡΎΚΥΘ§ΦΧΕχ Βœ÷Ε‘œΗΑϊΒΡ…χΆΗΓΘ
≥ΐΝΥ…œ ω‘ωΦ”πΛ“Κ…χΆΗΒΡΖΫΖ®Θ§ΜΙ”–“Μ÷÷Ηϋ÷±Ϋ”ΒΡΑλΖ®Θ§ΨΆ «»ήΫβπΛ“ΚΓΘΫΪΆΗΟς÷ ΥαΟΗ(hyaluronidase)ΓΔN- ““θΘΜυ -L- ΑκκΉΑ±Υα (NAC) ΚΆΡΨΙœΒΑΑΉΟΗ (papain) ΧμΦ”ΒΫ÷ΤΦΝ÷–Μρ–ό ΈΒΫΡ…ΟΉΝΘ±μΟφΘ§ΡήΤπΒΫ»ήΫβπΛ“ΚΒΡ–ßΙϊΘ§≤Δ«“ ‘―ι ΐΨί÷ΛΟςΦ”»κ’β–©πΛ“ΚΥ°ΫβΦΝ÷°ΚσΘ§≤ΜΜαΕ‘–Γ≥Π…œΤΛœΗΑϊ≤ζ…ζ”ΑœλΘ§ΒΪ»¥Ρή”––ß‘ωΦ”“©ΈοΒΡ…χΆΗ”κΈϋ ’ [26ΓΣ27]ΓΘ
Ήέ…œΥυ ωΘ§ΡΩ«ΑΧαΗΏ“©ΈοΕ‘πΛ“ΚΜρπΛΡΛ…χΆΗΒΡ÷ς“ΣΖΫΖ®Ω…“‘Η≈ά®ΈΣ ΘΚ‘ωΦ””κπΛΡΛΒΡΫ”¥Ξ ±Φδ”κΨ÷≤Ω“©Έο≈®Ε» ( πΛΡΛΈϋΗΫ )ΓΔΧαΗΏ“©Έο‘ΎπΛ“Κ÷–ΒΡά©…Δ ( πΛΡΛ…χΆΗ ) “‘ΦΑΨ÷≤Ω¥ρΩΣπΛΡΛ≤ψ ( πΛ“ΚΥ°Ϋβ )ΓΘ
3.3 ≥Π…œΤΛœΗΑϊΤΝ’œ
ΡΩ«Α¥σΕύ ΐΒΡ―–ΨΩΕΦΦ·÷–‘Ύ»γΚΈΆΜΤΤ≥Π…œΤΛœΗΑϊΤΝ’œΘ§œ÷”–ΒΡ Βœ÷œΗΑϊ…χΆΗΒΡΦΦ θ÷ς“ΣΈΣΕύκΡ–ό Έ ( Μ·―ßΗΡΙΙ )ΓΔΗ®ΝœΧμΦ” ( »γ…χΆΗ¥ΌΫχΦΝ )ΓΔΗΡ±δΦΝ–Ά ( Ρ…ΟΉΝΘΓΔΈΔ»ι ) Β»Θ§Μρ’β–©ΦΦ θΒΡΝΣΚœ Ι”ΟΓΘ
3.3.1 ΕύκΡΒΡΜ·―ß–ό Έ
ΫœΗΏΒΡ logP ÷Β”–÷ζ”Ύ“©Έο¥©ΆΗœΗΑϊΡΛΒΡΝΉ÷§ΥΪΖ÷Ή”≤ψ [28]ΓΘ“ρ¥ΥΘ§Ά®ΙΐΜ·―ß–ό ΈΧαΗΏΕύκΡΒΡ«Ή÷§–‘Ρή‘ω«ΩΤδœΗΑϊ…χΆΗΡήΝΠΓΘΡΩ«ΑΘ§ΧαΗΏΕύκΡ«Ή÷§–‘ΒΡΜ·―ßΖΫΖ®ΈΣ÷§÷ Μ·–ό ΈΘ§”÷Ω…œΗΖ÷ΈΣΚœ≥…«Α“©”κΖ««Α“© 2 ÷÷Θ§Εΰ’ΏΒΡ÷ς“Σ«χ±π‘Ύ”Ύ «Ζώ–η“ΣΆ®Ιΐ¥ζ–Μ ΆΖ≈≥ωΜν–‘“©ΈοΘ§±ΨΈΡΕ‘¥Υ≤Μ”ηœΗΖ÷ΓΘPARMENTIER Β»άϊ”ΟΕΰΝρΦϋΫΪΒΆΖ÷Ή”Ω«ΨέΧ«”κΑ§»ϊΡ«κΡΫαΚœΘ§œ‘÷χΧαΗΏΝΥΑ§»ϊΡ«κΡΒΡ«Ή÷§–‘ΦΑΩΎΖΰΫΒΧ«–ßΙϊ [29]ΓΘ÷§ΖΨΥα–ό Έ «”Π”ΟΫœΙψΖΚΒΡΧαΗΏΕύκΡ«Ή÷§–‘ΒΡΖΫΖ®Θ§Τδ÷–ΒΡΒδ–ΆΑΗάΐ «ΩΎΖΰΥς¬ξ¬≥κΡ÷ΤΦΝΒΡ≥…ΙΠΩΣΖΔΓΘ»ΜΕχΘ§≤ΔΖ«Υυ”–ΒΡ÷§ΖΨΥα–ό ΈΕΦΡήΤπΒΫ‘ωΦ”ΕύκΡΩΎΖΰΈϋ ’ΒΡ–ßΙϊΓΘTRIER Β»Ζ÷±πΫχ––ΝΥΥς¬ξ¬≥κΡ”κάϊά≠¬≥κΡΒΡΩΎΖΰΗχ“© ‘―ιΘ§‘ΎœύΆ§¥ΠΖΫΒΡ«ιΩωœ¬Θ§ΩΎΖΰΥς¬ξ¬≥κΡΉι≥ωœ÷ΝΥΟςœ‘ΒΡΈϋ ’–ßΙϊΘ§άϊά≠¬≥κΡ‘ρΦΗΚθΟΜ”–Έϋ ’Θ§’βΥΒΟς≤ΔΖ«Υυ”–ΒΡ÷§÷ Μ·ΕΦΡήΧαΗΏΕύκΡΒΡ…χΆΗ–‘ΓΘ‘ΎΕύκΡ¥©ΆΗœΗΑϊΒΡΙΐ≥Χ÷–¥φ‘ΎΉ≈“Μ÷÷ΈΔΟνΒΡΤΫΚβΘ§Φ»“Σ±Θ÷Λ≥δΉψΒΡ”κœΗΑϊΡΛΦδΒΡœύΜΞΉς”ΟΘ§Ά§ ±”÷≤ΜΡή”…”ΎΡΛ≤ε»κ≥ΧΕ»Ιΐ«ΩΕχΉηΑ≠ΕύκΡΒΡΩγœΗΑϊΉΣ‘Υ [30]ΓΘΕ‘”ΎΥς¬ξ¬≥κΡά¥ΥΒΘ§Τδ≤ύΝ¥ΫαΙΙΈΣΕύκΡ”κœΗΑϊΡΛΒΡΫαΚœΧαΙ©ΝΥΚœ ΒΡΤΫΚβΒψΘ§ΜΜ―‘÷°Θ§”––©«Ή÷§Μ·–ό Έ≤Δ≤Μ“ΜΕ®ΜαΧαΗΏΕύκΡ…χΆΗΒΡΡήΝΠ [11]ΓΘ
Μ·―ß–ό Έ≥ΐΡή‘ω«ΩΕύκΡΒΡ«Ή÷§–‘ΆβΘ§ΜΙΡήΆ®Ιΐ–ό ΈΒΡ≈δΧε Βœ÷ ήΧεΜρΉΣ‘ΥΧεΫιΒΦΒΡΩγΡΛ‘Υ δΘ§¥”ΕχΧαΗΏΕύκΡΒΡΩγΡΛ…χΆΗ–߬ ΓΘΕύ÷÷≈δΧε“―±Μ≥…ΙΠΒΊ”Π”Ο‘ΎΕύκΡΒΡ–ό Έ≤Ώ¬‘÷–Θ§»γΉΣΧζΒΑΑΉ (Tf)ΓΔΈ§…ζΥΊ B12(VB12)ΓΔ…ζΈοΥΊΓΔ»Ξ―θΒ®ΥαΓΔΧ«άύΦΑœΗΑϊ¥©ΆΗκΡ (CPP) Β»ΓΘTf «ΗΚ‘πΧεΡΎΧζΉΣ‘ΥΒΡΒΑΑΉ÷ Θ§SHAH Β»ΫΪ Tf Ά®ΙΐΕΰΝρΦϋ–ό ΈΒΫ“»ΒΚΥΊ…œΘ§‘ΎΧεΆβ¥©ΆΗ Caco-2 œΗΑϊ≤ψΒΡ ‘―ι÷–Ιέ≤λΒΫ“»ΒΚΥΊ -Tf Η¥ΚœΈοΡήάϊ”Ο Tf ήΧε (TfR) Άξ≥……χΆΗΘ§±Μ…ψ»ΓΒΡ“»ΒΚΥΊΝΩ±»Έ¥–ό Έ“»ΒΚΥΊΉιΧαΗΏΝΥ 5 ΓΪ 15 ±Ε [31]ΓΘΈΡœΉ±®ΒάΘ§Έ§…ζΥΊ–ό ΈΒΡΕύκΡΡήάϊ”ΟΈ§…ζΥΊΉΣ‘ΥΆ®¬ΖΆξ≥…ΕύκΡΒΡΉΣ‘Υ [32ΓΣ33]ΓΘΫΪ VB12 Ζ÷±πΝ§Ϋ”ΒΫ“»ΒΚΥΊΦΑ“»ΗΏ―ΣΧ«ΥΊ―υκΡ -1(GLP-1) …œΘ§ΩΎΖΰΗχ“©ΚσΕΦ»ΓΒΟΝΥΫœΚΟΒΡΫΒΧ«–ßΙϊΘ§÷ΛΟςΝΥΗΟ–ό ΈΖΫΑΗΒΡΩ…–––‘ΓΘ‘ΎΩ’≥ΠΚΆΜΊ≥Π…œΤΛœΗΑϊ…œΙψΖΚ¥φ‘ΎΉ≈…ζΈοΥΊΦΑΒ®Υα ήΧεΘ§“ρ¥ΥΆ®Ιΐ‘ΎΕύκΡ…œ–ό Έ…ζΈοΥΊΜρΒ®ΥαΨΆΡήάϊ”ΟΡΤάκΉ”“άάΒ–‘ΉΣ‘ΥΧεΫιΒΦΒΡ…χΆΗΉς”ΟΘ§Άξ≥…ΕύκΡΈϋ ’ [34]ΓΘYOUN Β»ΫΪ…ζΈοΥΊγζγξθΘ―«ΑΖΜνΜ·θΞbiotin-NHS) –ό ΈΒΫ GLP-1 ΒΡ Lys24 ΈΜΒψ…œΘ§≤ΜΫωΧαΗΏΝΥΕύκΡΒΡ«Ή÷§–‘Θ§Ά§ ±“≤άϊ”ΟΉΣ‘ΥΧε Βœ÷ΝΥΕύκΡΒΡ÷ςΕ·ΉΣ‘ΥΘ§’β÷÷–≠Ά§Ής”Οœ‘÷χ‘ωΦ”ΝΥ GLP-1 ‘ΎΧεΡΎΒΡΈϋ ’ [35]ΓΘΧεΆβ ‘―ι÷ΛΟςΘ§Ι≤ΦέΝ§Ϋ”»Ξ―θΒ®ΥαΒΡΒΆΖ÷Ή”ΗΈΥΊΕ‘ΡΤάκΉ”ΫιΒΦΒΡΒ®ΥαΉΣ‘ΥΧε”–ΫœΚΟΒΡ―Γ‘ώ–‘Θ§‘Ύ¥σ σΚΆΚοΧεΡΎΒΡΩΎΖΰ…ζΈοάϊ”ΟΕ»¥οΒΫ33.5ΘΞ”κ 19.9ΘΞ [36ΓΣ37]ΓΘCPP ΨΏ”–ΧΊ βΒΡΑ±ΜυΥα–ρΝ–Θ§Ρή”––ß‘ωΦ”…ζΈο“©ΈοΒΡ≥ΠΒάΈϋ ’Θ§ΒΪΤδ…χΆΗΜζ÷Τ…–≤Μ °Ζ÷«ε≥ΰΓΘKRISTENSEN Β»άϊ”ΟΒΑΑΉ»ΎΚœ ÷ΕΈΘ§≥…ΙΠΫΪ“ΜœΒΝ– CPP ±μ¥ο‘Ύ PTH( 1-34) ΒΡΡ©ΕΥ…œΘ§≤ΔΆ®Ιΐ―–ΨΩ±μΟςΫΪ CPP ΒΡκΡΕΈΝ§Ϋ”ΒΫ N ΕΥΡή≤ζ…ζ±»Ν§Ϋ”ΒΫ C ΕΥΗϋ«ΩΒΡœΗΑϊ…χΆΗΉς”ΟΘ§ΒΪ CPPΒΡΦ”»κΜα”Αœλ PTH(1-34) ΒΡΕΰΦΕΫαΙΙΘ§¥”Εχ”ΑœλΕύκΡΒΡ…ζΈοΜν–‘ [38]ΓΘ
≥Θ”Ο≈δΧε“‘ΦΑœύ”ΠΉΣ‘ΥΧεΒΡΉήΫα»γ±μ1Υυ Ψ[39]ΓΘ

Ήέ…œΥυ ωΘ§ΕύκΡΒΡΜ·―ß–ό ΈΡΩ«Α÷ς“Σ¥”‘ωΦ”«Ή÷§–‘ΧαΗΏ±ΜΕ·ά©…Δ“‘ΦΑ–ό Έ≈δΧε‘ωΦ”÷ςΕ·ΉΣ‘ΥΝΫΖΫΟφΫχ––―–ΨΩΘ§ΗΟΦΦ θΒΡΒΞΕά”Π”ΟΆυΆυ≤Δ≤ΜΡή¥οΒΫάμœκΒΡΧεΡΎΈϋ ’–ßΙϊΘ§–η“Σ”κΤδΥϊ¥Ό…χΖΫΖ®ΝΣΚœ”Π”ΟΓΘ
3.3.2 …χΆΗ¥ΌΫχΦΝΒΡ”Π”Ο
…χΆΗ¥ΌΫχΦΝΡή”––ßΧαΗΏΕύκΡ“©ΈοΒΡ…χΆΗ–ßΙϊΘ§ΡΩ«Α“―”–“Μ–©ΕύκΡ“©Έοάϊ”ΟΗΟΦΦ θ Βœ÷ΝΥΩΎΖΰΗχ“©Θ§≤Δ“―ΩΣ’ΙΝΌ¥≤ ‘―ιΘ§»γ“»ΒΚΥΊΓΔGLP-1ΓΔΫΒΗΤΥΊΓΔPTH Β»ΓΘ…χΆΗ¥ΌΫχΦΝΒΡ…χΆΗ‘≠άμΩ…ΉήΫαΈΣ“‘œ¬ 3 ÷÷ΘΚΔΌ¥ρΩΣœΗΑϊΦδΒΡΫτΟήΝ§Ϋ”Θ§¥ΌΫχΕύκΡΆ®ΙΐœΗΑϊ≈‘¬Ζ…χΆΗ ΘΜΔΎ‘ωΦ”œΗΑϊΡΛΒΡΝςΕ·–‘Θ§άϊ”Ύ“©Έο¥©ΆΗœΗΑϊΡΛΘ§¥”Εχ Βœ÷“©ΈοΒΡ…χΆΗ ΘΜΔέ…œ ω 2 ÷÷Μζ÷ΤΒΡΙ≤Ά§Ής”Ο [40]ΓΘΜυ”Ύ“‘…œ‘≠άμΘ§ΡΩ«Α≥Θ”ΟΒΡ…χΆΗ¥ΌΫχΦΝ÷ς“ΣΑϋά®ΘΚ±μΟφΜν–‘ΦΝΓΔΗΤάκΉ”ρϋΚœΦΝΓΔ÷–Ν¥÷§ΖΨΥαΓΔΒ®―ΈΦΑœΗΨζΕΨΥΊΓΘ…χΆΗ¥ΌΫχΦΝ”κΕύκΡ“©ΈοΙ≤Μλ÷Τ±Η≥…ΒΡ≥Θ”ΟΙΧΧε÷ΤΦΝ“≤Ω…¥οΒΫ¥Ό…χΡΩΒΡΘ§“ρ¥Υ ήΒΫΝΥΤσ“ΒΒΡΙψΖΚΙΊΉΔΓΘ
ΫΒΒΆ≥Π…œΤΛœΗΑϊΆβΒΡΗΤάκΉ”≈®Ε»Ρή»≈¬“ΑϊΦδΫτΟήΝ§Ϋ”÷–ΒΡΗΤάκΉ”“άάΒ–‘πΛΉ≈ΒΑΑΉΘ§¥”Εχ¥ρΩΣΫτΟήΝ§Ϋ”Θ§‘ωΦ”ΕύκΡ“©ΈοΒΡ…χΆΗΓΘΗΤάκΉ”ρϋΚœΦΝΨΆ «ΗυΨί’β“Μ‘≠άμ”Π”Ο”Ύ¥ΌΫχΕύκΡ“©Έο…χΆΗΒΡ―–ΨΩΘ§EDTA «Τδ÷–Ήν≥Θ”ΟΒΡΗΤάκΉ”ρϋΚœΦΝΓΘOramed ΙΪΥΨ‘ΎΑ§»ϊΡ«κΡ÷–Φ”»κ…χΆΗ¥ΌΫχΦΝ EDTA ”κ¥σΕΙΒΑΑΉΟΗ“÷÷ΤΦΝ÷Τ±Η≥Π»ή÷ΤΦΝΘ§ Βœ÷ΝΥΑ§»ϊΡ«κΡΒΡΩΎΖΰΈϋ ’Θ§≤Δ‘Ύ Beagle »°ΡΘ–Ά÷–»ΓΒΟΝΥΟςœ‘ΒΡΩΎΖΰΫΒΧ«–ßΙϊ [41]ΓΘ“Μ–©œΗΨζΕΨΥΊΡήΧΊ“λ–‘ΒΊΤΤΜΒΫτΟήΝ§Ϋ”÷–ΒΡπΛΉ≈ΒΑΑΉ (E-cadherin) ΦΑΟή±’ΒΑΑΉ (claudine) Β»Θ§“‘‘ωΦ”ΕύκΡΕ‘…œΤΛœΗΑϊ≤ψΒΡ…χΆΗΘ§»γ≤ζΤχΦ‘ΡΛΗΥΨζ≥ΠΕΨΥΊ (CPE)[40]ΓΘFASANO Β»ΫΪΖβ±’–Γ¥χΕΨΥΊ(ZOT) ”Π”Ο”Ύ“»ΒΚΥΊΩΎΖΰ÷ΤΦΝΒΡ…ηΦΤΘ§ΫαΙϊ÷ΛΟςΦ”»κ ZOT œ‘÷χ‘ω«ΩΝΥ“»ΒΚΥΊΒΡ…χΆΗ–߬ Θ§±»≤ΜΦ”ZOT ΒΡΕ‘’’ΉιΧαΗΏΝΥ 10 ±ΕΘ§≤Δ«“Ρή±Θ≥÷–Γ≥Π…œΤΛœΗΑϊ≤ψΒΡΆξ’ϊ–‘ [42]ΓΘΒΪ¥ρΩΣΫτΟήΝ§Ϋ”Ω…ΡήΜαΒΦ÷¬“Μ–©÷¬≤ΓΈΔ…ζΈο“≤ΗζΥφΉ≈ΕύκΡ“©ΈοΫχ»κ―Σ“Κ―≠ΜΖ‘λ≥…»Ϊ…μΗ–»ΨΘ§“ρ¥ΥΨΏ”–“ΜΕ®ΒΡΑ≤»Ϊ–‘Ζγœ’ΓΘ
≥ΐΆ®ΙΐΑϊΦδ…χΆΗΆΨΨΕΆβΘ§”–ΚήΕύ…χΆΗ¥ΌΫχΦΝ «Ά®ΙΐΩγœΗΑϊΉΣ‘ΥΆΨΨΕΤπΉς”ΟΒΡΓΘ±μΟφΜν–‘ΦΝΉςΈΣ“Μ÷÷…χΆΗ¥ΌΫχΦΝΘ§÷ς“ΣΆ®Ιΐ‘ωΦ”Ε‘œΗΑϊΡΛΒΡ»≈Ε·ΓΔΗΡ±δœΗΑϊΡΛΒΡΝςΕ·–‘Θ§¥”Εχ Βœ÷ΕύκΡ“©ΈοΩγΡΛ…χΆΗΘ§Τδ÷–”Π”ΟΫœΙψΖΚΒΡ «ΙοΥαΡΤ (C10)ΓΘΙοΥαΡΤ «“Μ÷÷“θάκΉ”±μΟφΜν–‘ΦΝΘ§MerLion Pharma ΙΪΥΨ
ΫΪ GIPET® ΦΦ θ”Π”ΟΒΫΩΎΖΰ¥σΖ÷Ή”…ζΈο“©ΈοΒίΥΆ÷–Θ§ΡΩ«Α“― Βœ÷ΝΥ“»ΒΚΥΊΦΑ GLP-1 άύΥΤΈοΒΡΒίΥΆ [43]ΓΘΕ‘”ΎΙοΥαΡΤΒΡ¥Ό…χΜζ÷ΤΘ§“Μ÷±“‘ά¥ΕΦ”–Ϋœ¥σ’υ“ιΘ§Τ’±ι±Μ»œΆ§ΒΡ « ΘΚΒΆ≈®Ε»ΙοΥαΡΤΡή¥ρΩΣΫτΟήΝ§Ϋ”Θ§ Βœ÷“©ΈοΒΡΑϊΦδ‘Υ δ ΘΜΕχΗΏΦΝΝΩΙοΥαΡΤ‘ρ÷ς“Σ «ΗΡ±δΡΛ…χΆΗ–‘Θ§Β±¥σΦΝΝΩΙοΥαΡΤ±©¬Ε”Ύ…œΤΛœΗΑϊ ±Θ§œΗΑϊΡΛΜα≤ζ…ζΈ¬ΚΆ«“Ω…ΡφΒΡ»≈Ε·Θ§–≠÷ζ“©ΈοΆξ≥…ΑϊΡΎΉΣ‘Υ [44]ΓΘΗυΨίΈΡœΉ±®Βά [43ΓΣ44]Θ§ΒΆΖ÷Ή”ΗΈΥΊΦΑ»ΞΑ±Φ”―ΙΥΊΖ÷±π”κΙοΥαΡΤΉιΚœ”Π”ΟΘ§Ήν÷’Ω…¥οΒΫ 3.9ΘΞΦΑ 2.4ΘΞΒΡΩΎΖΰ…ζΈοάϊ”ΟΕ»ΓΘ2019 Ρξ Novo NordiskΙΪΥΨΙΪ≤ΦΝΥ“ΜœνΩΎΖΰ≥Λ–ß“»ΒΚΥΊΒΡΔρΤΎΝΌ¥≤ ‘―ι±®ΗφΘ§ΗΟ≤ζΤΖ”Π”Ο GIPET® ΦΦ θΘ§ Βœ÷ΝΥ 1.5ΘΞΓΪ 2.0ΘΞΒΡ ΩΎ Ζΰ …ζ Έο άϊ ”Ο Ε»Θ§ «“ ”– Ϋœ ΗΏ ΒΡ Α≤ »Ϊ –‘ [45]ΓΘEmisphere ΙΪΥΨΩΣΖΔΝΥ“ΜΩνΩΎΖΰ¥σΖ÷Ή”ΒίΥΆΦΦ θΓΣΓΣEligen™Θ§Χα≥ω“ΜœΒΝ–“‘÷–Ν¥÷§ΖΨΥαΈΣΜυ¥ΓΒΡ…χΆΗ¥ΌΫχΦΝΘ§Αϋά® SNACΓΔ8-(2- τ«Μυ±ΫΦΉθΘΑΖΜυ )-ΙοΥαΡΤ (SNAD)ΓΔ8-[N-(2- τ«Μυ -4- ΦΉ―θΜυ±ΫΦΉθΘΜυ ) Α±Μυ ] –ΝΥαΡΤ (4-MOAC)ΓΔ8-[N-(2- τ«Μυ -5-¬»±ΫΦΉθΘΜυ ) Α±Μυ ] –ΝΥαΡΤ ( 5-CNAC)ΓΔ4-[ ( 4-¬» -2- τ«Μυ±ΫΦΉθΘΜυ ) Α±Μυ ] ΕΓΥαΡΤ (4-CNAB) Β»Θ§Τδ÷– SNAC ”Π”ΟΫœΙψΖΚΓΘΡΩ«ΑΘ§ΫœΈΣ¥σΦ“Ϋ” ήΒΡΙΊ”Ύ SNAC ΒΡ¥Ό…χΜζ÷Τ « ΘΚSNAC Ά®ΙΐΖ«Ι≤ΦέΦϋΉς”Ο”κ…ζΈο¥σΖ÷Ή”–Έ≥… ηΥ°–‘ ( «Ή÷§–‘ ) Η¥ΚœΈοΘ§¥”Εχœ‘÷χΧαΗΏΕύκΡ“©ΈοΒΡ…χΆΗ–‘ [46]ΓΘΆ§ ±Θ§“≤”–»Υ»œΈΣ SNAC Ρή‘ωΦ”œΗΑϊΡΛΒΡΝςΕ·–‘Θ§ΦΧΕχΧαΗΏΕύκΡ“©ΈοΒΡ…χΆΗΓΘSNAC “―±Μ≥…ΙΠ”Ο”Ύ“»ΒΚΥΊΓΔGLP-1 άύΥΤΈοΓΔPTHΓΔΫΒΗΤΥΊΦΑ…ζ≥ΛΦΛΥΊΒ»―–ΨΩ÷–Θ§Τδ÷–≥…ΙΠ…œ –ΒΡ≤ζΤΖΈΣΩΎΖΰΥς¬ξ¬≥κΡΤ§ ( ΩΎΖΰ…ζΈοάϊ”ΟΕ»ΈΣ 1.5ΘΞ )ΓΘ“‘…ΪΝ–Α≤ΧΊά≠±¥≈ΖΙΪΥΨ÷Τ±ΗΝΥ“ΜΩνΚ§”–PTHΓΔSNAC ”κ“»ΒΑΑΉΟΗ“÷÷ΤΦΝΒΡ≥Π»ήΤ§Θ§ΝΌ¥≤ΔώΤΎ ‘―ι÷–”– 42 ΈΜ÷Ψ‘Η’ΏΒΡΧεΡΎ“©Ε·―ßΫαΙϊ÷ΛΟςΩΎΖΰPTH Τ§ΦΝ (1.8 mg) ”κΤΛœ¬ΉΔ…δ PTH »ή“Κ (20 µg)Ρή≤ζ…ζœύΆ§ΒΡ cmax ÷ΒΘ§ΒΪΩΎΖΰΆΨΨΕΒΡ PTH «ε≥ΐΗϋΩλ [47]ΓΘΆ§ ±‘ΎΤ§ΦΝ÷–‘ωΦ”ΩΙΥαΦΝΧΦΥα«βΡΤΡή‘ωΦ”35ΘΞΒΡ“©ΈοΈϋ ’ΓΘ
Ήέ…œΥυ ωΘ§”…”Ύ÷Τ±ΗΙΛ“’ΦρΒΞΓΔ¥Ό…χΙΠΡήΫœΚΟΓΔΙΛ“’Ζ≈¥σ»ί“ΉΒ»”≈ΒψΘ§ΡΩ«Α…χΆΗ¥ΌΫχΦΝ“―ΒΟΒΫΙψΖΚ―–ΨΩΘ§œύ–≈Έ¥ά¥ΫΪΜα”–ΗϋΕύΒΡ…ζΈο¥σΖ÷Ή””–ΆϊΆ®ΙΐΗΟΦΦ θ Βœ÷ΩΎΖΰΗχ“©ΓΘ
3.3.3 Ρ…ΟΉΒίΥΆ‘ΊΧεΒΡ”Π”Ο
…χΆΗ¥ΌΫχΦΝ‘ωΦ”…ζΈοάϊ”ΟΕ»ΒΡΉς”Ο”–œόΘ§ΡΩ«ΑΫœΡ―Ά®ΙΐΗΟΖ®¥οΒΫ 10ΘΞ“‘…œΒΡ…ζΈοάϊ”ΟΕ»Θ§“ρ¥ΥΩΤ―–»Υ‘±ΫΪΙΊΉΔΒψΖ≈‘ΎΝΥΡ…ΟΉ‘ΊΧε…œΘ§œΘΆϊΡήάϊ”ΟΡ…ΟΉ‘ΊΧεΧαΗΏΕύκΡΒΡΈϋ ’ΓΘΡ…ΟΉ‘ΊΧεΧαΗΏΕύκΡΈϋ ’ΒΡ”≈ Τ‘Ύ”ΎΘΚΔΌΡ…ΟΉΝΘΡήΉη÷ΙΈΗ≥ΠΒάΟΗΕ‘ΕύκΡΒΡΫΒΫβΘ§ΤπΒΫ±ΘΜΛΕύκΡΒΡΉς”Ο ΘΜΔΎΆ®ΙΐΒς’ϊΝΘΨΕΓΔΒγΚ…ΦΑ±μΟφ–‘÷ Β» ÷ΕΈΘ§Ω…‘ωΦ”ΈΗ≥ΠΒά…œΤΛœΗΑϊΕ‘ΝΘΉ”ΒΡ…ψ»Γ ΘΜΔέΕ‘ΝΘΉ”±μΟφΫχ––÷ςΕ·Α–œρ≈δΧεΒΡ–ό ΈΘ§Ρή ΙΝΘΉ””κœΗΑϊΡΛ±μΟφΒΡ ήΧεΜρΉΣ‘ΥΧεΧΊ“λ–‘ΫαΚœΘ§¥”ΕχΆξ≥… ήΧεΫιΒΦΒΡΩγœΗΑϊΡΛΉΣ‘ΥΘ§≤Δ«“”κ÷±Ϋ”ΫΪ≈δΧε–ό Έ‘Ύ“©Έο…œΒΡΖΫΖ®œύ±»Θ§≤Μ¥φ‘ΎΫΒΒΆΕύκΡΜν–‘ΒΡΖγœ’ ΘΜΔήΆ®ΙΐΕ‘Ρ…ΟΉΝΘΒΡ÷Τ±ΗΙΛ“’ΦΑ‘ΊΧε≤ΡΝœΒΡ”≈Μ·Θ§Ω…ΩΊ÷Τ“©ΈοΒΡ ΆΖ≈ΈΜ÷ΟΦΑ ΆΖ≈ΥΌ¬ Θ§¥”Εχ”–÷ζ”Ύ“©ΈοΒΡΕ®ΈΜΓΔΕ®ΝΩ ΆΖ≈ [ 48]ΓΘΜυ”Ύ«ΑΈΡΥυ ωΩγ…œΤΛœΗΑϊΉΣ‘ΥΒΡ 5 ΗωΆΨΨΕΘ§Ρ…ΟΉ‘ΊΧε Βœ÷“©ΈοΈϋ ’÷ς“ΣΆ®Ιΐ“‘œ¬ 3 ÷÷ ÷ΕΈ ΘΚΔΌάϊ”Ο‘ΊΧε≤ΡΝœΒΡ«Ή÷§ΧΊ–‘Θ§‘ω«Ω”κœΗΑϊΡΛΒΡ«ΉΚΆΝΠ ΘΜΔΎάϊ”ΟΗς÷÷≈δΧε (TfΓΔVB12ΓΔΒ®ΥαΒ» )ΓΔCPP ΦΑ Fc Τ§ΕΈΒ»Ε‘‘ΊΧε±μΟφΫχ––÷ςΕ·Α–œρ–ό ΈΘ§ΧαΗΏ÷ςΕ·ΉΣ‘Υ–ß¬ ΘΜΔέάϊ”Ο…œΤΛœΗΑϊ≤ψ…œΒΡΧΊ βœΗΑϊ ( »γ M œΗΑϊΜρ±≠Ή¥œΗΑϊ ) Ϋχ––÷ςΕ·ΉΣ‘ΥΓΘ
÷§άύ≤ΡΝœ÷Τ≥…ΒΡΡ…ΟΉΝΘ÷ς“ΣΑϋά®ΙΧΧε÷§÷ Ρ…ΟΉΝΘ (SLN) ΦΑ÷§÷ ΧεΘ§“ρ¥Υάϊ”Ο’β 2 ÷÷Ρ…ΟΉ‘ΊΧεΫχ––ΕύκΡ“©ΈοΒίΥΆΒΡ―–ΨΩΫœΕύΓΘ÷§÷ ΧεΉςΈΣ“Μ÷÷Ψ≠Βδ‘ΊΧεΘ§“≤±Μ”Π”ΟΒΫΩΎΖΰΕύκΡ“©ΈοΒίΥΆΒΡ―–ΨΩ÷–ΓΘDiasome ΙΪΥΨ…ηΦΤΝΥ“ΜΩνΗΈΑ–œρΒΡ“»ΒΚΥΊ÷§÷ Χε(HDV-1)Θ§œΒΫΪ“»ΒΚΥΊΑϋ‘ΊΫχ»κΝΘΨΕ–Γ”Ύ 150 nmΒΡ÷§÷ Χε÷–Θ§≤Δ‘Ύ±μΟφΫχ––Α–œρ–ό ΈΘ§ ΙΤδΡή÷±Ϋ”Α–œρΗΈ‘ύΘ§¥”Εχ÷ΊΫ®“»ΒΚΥΊ‘ΎΗΈ‘ύ≤ΩΈΜΒΡ’ΐ≥Θ…ζάμœλ”ΠΘ§“‘¥οΒΫΩΎΖΰΫΒΧ«–ßΙϊ [49]ΓΘKISEL Β»ΩΣΖΔΝΥ“ΜΩν“‘ΕΰΉΊιΒθΘΝΉ÷§θΘ““¥ΦΑΖ (DPPE) ΈΣ≤ΡΝœΒΡ÷§÷ ΧεΘ§ΗΟ≤ΡΝœ”κ¥ΪΆ≥ΒΑΜΤΝΉ÷§œύ±»ΨΏ”–ΗϋΗΏΒΡ»έΒψΘ§“ρ¥Υ÷Τ±ΗΒΡ÷§÷ ΧεΗϋΈ»Ε® ΘΜΧεΡΎ ‘―ι÷ΛΟςΗΟ÷§÷ ΧεΡή Βœ÷“»ΒΚΥΊΒΡ”––ßΒίΥΆΘ§ΨΏ”–ΩΎΖΰΫΒΧ«–ßΙϊ [50]ΓΘ
≥ΐ÷§÷ ΧεΆβΘ§Νμ“Μ÷÷ΨΏ”–¥ζ±μ–‘ΒΡ÷§άύ‘ΊΧεSLN “≤ ήΒΫΙψΖΚΙΊΉΔΓΘΒΪ SLN ÷ΜΕ‘ ηΥ°–‘“©ΈοΨΏ”–ΫœΗΏΒΡ‘Ί“©ΝΩΘ§“ρ¥Υ‘ΎΑϋ‘Ί«ΉΥ°–‘ΕύκΡ“©Έο ±ΨΆ“Σœ»ΫΪ“©Έο”κœύ”ΠΒΡ±μΟφΜν–‘ΦΝΆ®ΙΐΒγΚ…ΦδœύΜΞΉς”ΟΫαΚœΘ§–Έ≥… ηΥ°–‘Η¥ΚœΈοΘ§“‘ΧαΗΏΝΘΉ”ΒΡ‘Ί“©ΝΩΓΘCHEN Β»άϊ”ΟΗ¥»ιΖ®“‘”≤÷§ΥαΦΑΉΊιΒΥαΗ ”ΆθΞΈΣ‘≠Νœ÷Τ±ΗΝΥΫΒΗΤΥΊ SLNΘ§“©Ε·―ß ‘―ι÷ΛΟςΤδΩΎΖΰ…ζΈοάϊ”ΟΕ»ΗΏ¥ο 13ΘΞ [51]ΓΘCHEN Β»œ»ΫΪΑ§»ϊΡ«κΡ”κΒ®ΥαΡΤ÷Τ≥…ΫΚ χΘ§‘ΌΫΪΫΚ χΑϋΙϋ‘Ύ SLN ÷–Θ§’β―υΡή¥οΒΫ 97.7ΘΞΒΡΑϋΖβ¬ “‘ΦΑ 12ΘΞΒΡΩΎΖΰ…ζΈοάϊ”ΟΕ» [52]ΓΘ
¥ΥΆβΘ§ΜΙ”–ΚήΕύΧλ»ΜΜρΚœ≥…ΗΏΖ÷Ή”≤ΡΝœ±Μ”Ο”ΎΕύκΡ“©ΈοΩΎΖΰΒίΥΆΒΡ―–ΨΩΘ§»γΩ«ΨέΧ«ΓΔΚΘ‘εΥα―ΈΓΔΨέ»ιΥα - τ«Μυ““ΥαΙ≤ΨέΈο (PLGA)ΓΔΤœΨέΧ«Β» [53]ΓΘΤδ÷–Ω«ΨέΧ«±Μ÷ΛΟςΡή¥ρΩΣΫτΟήΝ§Ϋ”Θ§“ρ¥Υ”Π”ΟΙψΖΚΓΘ‘Ύ÷––‘ΧθΦΰœ¬Θ§Ω«ΨέΧ«œ‘’ΐΒγ–‘Θ§Ρή”κ¥χΗΚΒγΚ…ΒΡΗΏΖ÷Ή”–Έ≥…Ρ…ΟΉΝΘΓΘSONAJE Β»‘Ύ pH 6.0 œ¬ΫΪΩ«ΨέΧ«”κΨέ ΠΟ- Ι»Α±ΥαΜλΚœ–Έ≥…Ρ…ΟΉΝΘ”Οά¥ΗΚ‘Ί“»ΒΚΥΊΘ§Ε≥Η…ΚσΉΑ»κ≥Π»ήΫΚΡ“Θ§Ρή¥οΒΫ 20ΘΞΒΡΩΎΖΰ…ζΈοάϊ”ΟΕ» [54]ΓΘ“≤”–»ΥΕ‘Ω«ΨέΧ«Ρ…ΟΉΝΘ±μΟφΫχ––ΗΡ–‘Θ§ΫΪΗΤάκΉ”ρϋΚœΦΝ““Εΰ¥ΦΥΪ (2- Α±Μυ““ΜυΟ― ) ΥΡ““Υα(EGTA) –ό ΈΒΫΡ…ΟΉΝΘ±μΟφΘ§ΜρΆ®Ιΐ‘Ύ±μΟφ–ό ΈΝρ¥ΦΜυΆ≈“‘‘ωΦ”πΛΡΛΈϋΗΫ–‘Β» ÷ΕΈά¥¥ΌΫχΝΘΉ”Ψ≠œΗΑϊΦδΉΣ‘Υ [55]ΓΘΉή÷°Θ§Ω«ΨέΧ«ΉςΈΣ“Μ÷÷ΨΏ”–ΝΦΚΟ…ζΈοœύ»ί–‘ΒΡ≤ΡΝœΘ§”–Άϊ‘ΎΕύκΡ“©ΈοΩΎΖΰΒίΥΆ÷–ΖΔΜ”÷Ί“ΣΉς”ΟΓΘ≥ΐ…œ ωΡ…ΟΉΝΘΆβΘ§Nod ΙΪΥΨ…ηΦΤΝΥ“ΜΩνΨΏ”–…ζΈοπΛΗΫ–‘ΒΡΝΉΥαΗΤΡ…ΟΉΝΘΘ§¥ΠΖΫ÷–ΑϋΚ§Α§»ϊΡ«κΡΓΔΝΉΥαΗΤΦΑΒ®―ΈΜρ÷–Ν¥÷§ΖΨΥα (C8 ΓΪ C10)Θ§Ρή”––ß‘ωΦ”Α§»ϊΡ«κΡΒΡΩΎΖΰΈϋ ’Θ§ΡΩ«Α’ΐ‘ΎΫχ––ΔώΤΎΝΌ¥≤ ‘―ι [56]ΓΘ
‘ΎΡ…ΟΉΝΘ±μΟφΫχ––≈δΧε–ό ΈΘ§Ω…¥ΌΫχΡ…ΟΉΝΘΆ®Ιΐ÷ςΕ·ΉΣ‘ΥΆΨΨΕΫχ––Έϋ ’Θ§“≤ «―–ΨΩ»»Βψ÷°“ΜΓΘPRIDGEN Β»“‘Ψέ»ιΥα (PLA)-PEG ΈΣ≤ΡΝœ÷Τ±ΗΝΥΑϋ‘Ί“»ΒΚΥΊΒΡΡ…ΟΉΝΘΘ§≤Δ‘Ύ±μΟφ–ό ΈΝΥ IgG ΩΙΧεΒΡ FcΤ§ΕΈΘ§Ά®ΙΐΗΟΤ§ΕΈ”κ…œΤΛœΗΑϊ±μΟφΒΡ Fc ήΧε (FcRn)ΧΊ“λ–‘ΫαΚœΘ§ΫιΒΦ IgG ΩΙΧεΒΡΩγ…œΤΛœΗΑϊΉΣ‘ΥΘ§¥”Εχ Βœ÷ ήΧεΫιΒΦΒΡΡ…ΟΉΝΘ…ψ»Γ [57]ΓΘFAN Β»‘ΎΑϋ‘Ί“»ΒΚΥΊΒΡΩ«ΨέΧ« - Ψέ““¥ΦΥα (PGA) Ρ…ΟΉΝΘ±μΟφ–ό Έ»Ξ―θΒ®ΥαΘ§Ά®Ιΐ…œΤΛœΗΑϊ±μΟφΡΤάκΉ”“άάΒ–‘Β®ΥαΉΣ‘ΥΧεΆξ≥…ΕύκΡΉΣ‘Υ [58]ΓΘZHANG Β»÷Τ±ΗΝΥ¥χ”–…ζΈοΥΊΑ–ΆΖΒΡ÷§÷ ΧεΘ§≤Δ÷ΛΟςΗΟΡ…ΟΉΝΘΒΡΈϋ ’ «Ά®Ιΐ ήΧεΫιΒΦΒΡΡΎΆΧΆΨΨΕΆξ≥…ΒΡ [59]ΓΘ
‘Ύ–Γ≥ΠœΗΑϊ÷–Θ§≥ΐΝΥ’ΦΕύ ΐΒΡΥΔΉ¥…œΤΛœΗΑϊΆβΘ§ΜΙ”–“Μ–©ΨΏ”–ΧΊ βΙΠΡήΒΡœΗΑϊΘ§»γ M œΗΑϊΓΘM œΗΑϊ±μΟφΒΡΧ«ΒΑΑΉΫœ…ΌΓΔΥ°ΫβΟΗΜν–‘ΫœΒΆΘ§“ρ¥ΥΨΏ”–ΫœΗΏΒΡΩγœΗΑϊΡΛΉΣ‘ΥΡήΝΠΘ§Ω…ΉΣ‘ΥœΗΨζΓΔ≤ΓΕΨΓΔΩΙ‘≠ΦΑΡ…ΟΉΝΘΒ» [60]ΓΘΡΐΦ·ΥΊΨΏ”–ΫœΚΟΒΡ M œΗΑϊΑ–œρΡήΝΠΘ§ «“ΜάύΡή”κœΗΑϊΡΛ…œΒΡΧ«÷§ΜρΧ«ΒΑΑΉ…œΒΡΧ«ΜυΫχ––ΧΊ“λ–‘ΫαΚœΒΡΒΑΑΉ÷ ΓΘ‘ΎΡ…ΟΉΝΘ±μΟφΫχ––ΡΐΦ·ΥΊ–ό ΈΡή‘ωΦ” M œΗΑϊΕ‘ΝΘΉ”ΒΡ…ψ»ΓΓΘM œΗΑϊ±μΟφ¥φ‘ΎΒΡ“Μ–©≤Γ‘≠Χε Ε±π ήΧε“≤Ω…±Μ”ΟΉςΑ–±ξΘ§ Βœ÷Ε‘ M œΗΑϊΒΡΑ–œρΉς”ΟΓΘ≥ΐΑ–œρ M œΗΑϊΆβΘ§JINΒ»ΜΙ±®ΒάΝΥ“Μ÷÷ΡήΑ–œρ±≠Ή¥œΗΑϊΒΡΩ«ΨέΧ«Ρ…ΟΉΝΘΘ§ΗΟΡ…ΟΉΝΘ…œ–ό ΈΝΥ“ΜΕΈΑ–œρκΡ (CSK κΡ )Θ§‘ΎΤδΉς”Οœ¬ΡήΑ–œρ…œΤΛœΗΑϊ≤ψ÷–ΒΡ±≠Ή¥œΗΑϊΘ§άϊ”ΟΧΊΕ®œΗΑϊ Βœ÷“©ΈοΒΡΈϋ ’ [61]ΓΘ
3.3.4 –¬…χΆΗΦΦ θΒΡ”Π”Ο
“‘…œΫι…ήΒΡ «ΡΩ«Α≥Θ”Ο«“±ΜΙψΖΚ÷ΛΟς”––ßΒΡ“Μ–©ΦΦ θΓΘΥφΉ≈―–ΨΩΒΡ≤ΜΕœ…ν»κΘ§“Μ–©–¬ΦΦ θ“≤±Μ”Π”Ο”Ύ¥σΖ÷Ή”“©ΈοΒΡΒίΥΆ―–ΨΩΓΘάκΉ”»ή“Κ (ionic liquids) «“Μ÷÷Ϋω”…άκΉ”ΙΙ≥…ΓΔ»έΒψ–Γ”Ύ 100 ΓφΒΡ―ΈΓΘάκΉ”»ή“ΚΒΡ–Έ≥…‘≠άμάύΥΤ”ΎΙΧΧεΒΡ―ΈΚΆΙ≤ΨßΘ§ «Ά®ΙΐάκΉ”ΦϋΜρ«βΦϋΒ»Ής”ΟΝΠ–Έ≥…ΒΡ“Μ÷÷≥ΘΈ¬Μρ’ΐ≥ΘΧεΈ¬œ¬ΈΣ“ΚΧεΒΡ“Μ÷÷Έο÷ ΓΘ¥ΥΆβΘ§άκΉ”»ή“ΚΨΏ”–‘ωΦ”Ρ―»ή–‘“©Έο»ήΫβΕ»Β»Ής”ΟΘ§“ρ¥Υ“≤±ΜΙψΖΚ”Π”Ο”Ύ–ΓΖ÷Ή”“©ΈοΒΡΒίΥΆ [62ΓΣ63]ΓΘΫϋΡξά¥Θ§»ΥΟ«÷πΫΞΖΔœ÷ΝΥάκΉ”»ή“Κ‘ΎΒίΥΆ…ζΈο¥σΖ÷Ή”ΖΫΟφΒΡ«±ΝΠΓΘBANERJEE Β»άϊ”Ο”…Β®ΦνΚΆœψ“ΕΥα (CAGE)Ήι≥…ΒΡάκΉ”»ή“Κ÷Τ±ΗΝΥΩΎΖΰ“»ΒΚΥΊΗχ“©ΧεœΒΘ§ΧεΆβCaco-2 œΗΑϊΡΘ–Ά ‘―ιΥΒΟς CAGE άκΉ”»ή“ΚΕ‘“»ΒΚΥΊΒΡΒίΥΆΨΏ”–≈®Ε»“άάΒ–‘Θ§“ρ¥ΥΆΤΕœ «ΗΟ÷ΤΤΖΆ®Ιΐ¥ρΩΣΫτΟήΝ§Ϋ”ΓΔ‘ωΦ”ΑϊΦδΉΣ‘Υ Βœ÷ΒίΥΆ [64]ΓΘΆ§ ±ΧεΡΎ ‘―ι“≤ΥΒΟςΗΟ CAGE άκΉ”»ή“ΚΨΏ”–ΫœΚΟΒΡΫΒΧ«–ßΙϊΘ§«“ΩΎΖΰ 5 u/kg ΒΡ“»ΒΚΥΊΡή≤ζ…ζ”κΤΛœ¬ΉΔ…δ2 u/kg œύΥΤΒΡΈϋ ’–ßΙϊΓΘ
ΈΣ Βœ÷“»ΒΚΥΊΒΡΩγΡΛ‘Υ δΘ§Rani ΙΪΥΨΩΣΖΔΝΥ“ΜΩνΜυ”ΎΈΔ’κΦΦ θΒΡ÷«ΡήΒίΥΆœΒΆ≥ [15]ΓΘ’βΩν÷«ΡήΫΚΡ“ΨΏ”–“Μ÷÷ΓΑΤχ«ρΉ¥Γ±≥δΤχΫαΙΙΘ§Β±Ϋχ»κ–Γ≥ΠΚσΘ§Άβ≤ψ≥Π»ή“¬Ω«ΩΣ Φ»ήΫβΘ§¥ΞΖΔηέιΎΥαΚΆΧΦΥα«βΡΤΒΡΖ¥”ΠΘ§ ΆΖ≈≥ωΕΰ―θΜ·ΧΦΘ§ ΙΫΚΡ“ΡΎ≤ΩΫαΙΙ≈ρ’ΆΘ§ΫΪΈΔ’κ¥Χ»κΒΫ–Γ≥Π≥Π«Μ…œΘ§¥”Εχ Βœ÷“»ΒΚΥΊΒΡΩγ–Γ≥Π…œΤΛœΗΑϊ ΆΖ≈ΓΘ’β÷÷÷«ΡήΫΚΡ“÷±Ϋ”¥©ΆΗΝΥ≥Π±ΎΒΡΈοάμΤΝ’œΘ§“ρ¥ΥΒίΥΆ–߬ “≤ΒΟΒΫΫœ¥σΧαΗΏΘ§…ζΈοάϊ”ΟΕ»Ω…¥οΒΫ 50ΘΞ“‘…œΘ§≥…ΈΣ“Μ÷÷ΒίΥΆΕύκΡ“©ΈοΒΡ–¬ ΫΓΑΈδΤςΓ±ΓΘ
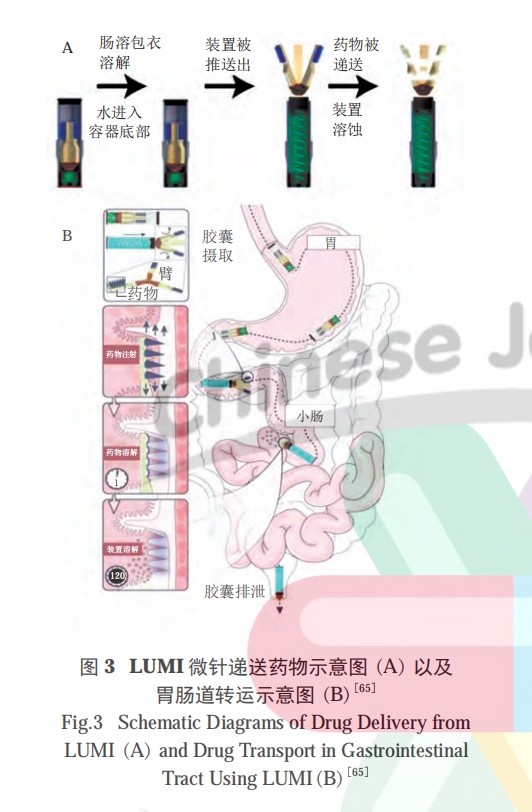
ABRAMSON Β»…ηΦΤΝΥ“ΜΩν«ΜΧεΩ……λ’ΙΒΡΈΔ’κΉΔ…δΤς (LUMI)Θ§ΗΟΈΔ’κΉΔ…δΤςΑϋΙϋ‘Ύ≥Π»ήΫΚΡ“÷–Θ§»γΆΦ 3 Υυ Ψ [65]ΓΘΒ±ΫΚΡ“Ϋχ»κΒΫ≥Π«ΜΚσΘ§ΫΚΡ“Ω«ΩΣ ΦΥ°ΫβΘ§Ά§ ±ΫΚΡ“άοΒΡ…χΆΗ±ΟΩΣ ΦΖΔΜ”Β·Μ…ΒΡΉς”ΟΘ§ΫΪ LUMI ΆΤ≥ωά¥Θ§¥Υ ±–«Ή¥ΒΡΈΔ’κΩΣ Φ…λ’ΙΘ§ΟΩ“ΜΗω…λ’≈ΩΣΒΡ±έΕΞ≤ΩΕΦ”–‘Ί“©ΈΔ’κΘ§’β–©ΈΔ’κάΈάΈ¥Χ»κ≥Π±ΎΘ§Ά®ΙΐΈΔ’κΒΡ¥©¥ΧΉς”ΟΫΪ“©Έο ΆΖ≈ΒΫ≥Π±ΎΆβ≤ύΘ§Άξ≥…“»ΒΚΥΊΒΡ ΆΖ≈ΓΘΗΟΈΔ’κ≤…”ΟΩ…ΫΒΫβ≤ΡΝœ÷Τ±ΗΘ§Ρή‘ΎΒί“©Ϋα χΚσ±ΜΫΒΫβΘ§“ρ¥ΥΨΏ”–ΫœΗΏΒΡΑ≤»Ϊ–‘ΓΘ”κΤΛœ¬ΉΔ…δœύ±»Θ§ΗΟΒίΥΆΧεœΒΡή¥οΒΫ 10ΘΞ“‘…œΒΡ…ζΈοάϊ”ΟΕ»Θ§“‘÷μΈΣΡΘ–ΆΒΡΕ·Έο ‘―ι÷ΛΟςΝΥΗΟ÷ΤΤΖΒΡΑ≤»Ϊ–‘ΓΘΗΟΒίΥΆΦΦ θΨΏ”–Τ’ –‘Θ§Έοά쥩¥ΧΈΔ’κ ”Ο”ΎΥυ”–άύ–ΆΒΡΕύκΡΘ§≤ΜΜα“ρΈΣΕύκΡ–‘÷ ≤ΜΆ§Εχ–η“ΣΒς’ϊ≤ΜΆ§ΒΡΗχ“© ÷ΕΈΘ§“ρ¥Υ“≤≥…ΈΣ“ΜΗωΫœ”–«±ΝΠΒΡΕύκΡ“©ΈοΒίΥΆΉΑ÷ΟΓΘ
4 Ϋα¬έ
œ÷”–ΒΡΧαΗΏΕύκΡ“©ΈοΩΎΖΰ…ζΈοάϊ”ΟΕ»ΒΡΦΦ θΜψΉήΦϊΆΦ 4ΓΘ

ΕύκΡ“©ΈοΩΎΖΰΒίΥΆΟφΝΌΉ≈÷ΎΕύΒΡ…ζΈοΤΝ’œΘ§Έό¬έ «ΥαΤΝ’œΓΔΟΗΤΝ’œΘ§ΜΙ «≥Π±ΎœΗΑϊ≤ψΤΝ’œΘ§ΡΩ«ΑΕΦ“―’“ΒΫΝΥΫœΚΟΒΡΫβΨω ÷ΕΈΓΘ»ΜΕχΡΩ«Α…œ –ΒΡΩΎΖΰΕύκΡ≤ζΤΖ”–œόΘ§¥σ≤ΩΖ÷―–ΨΩ»‘»Μ¥Π‘ΎΝΌ¥≤«ΑΜρΝΌ¥≤ ‘―ιΫΉΕΈΓΘΟΗ“÷÷ΤΦΝ”κ…χΆΗ¥ΌΫχΦΝΒΡΉιΚœ‘ΎΝΌ¥≤ ‘―ι÷–±Μ÷ΛΟς «“Μ÷÷”––ßΒΡΒίΥΆ ÷ΕΈΘ§ΒΪΗΟΖ®Ε‘”Ύ…ζΈοάϊ”ΟΕ»ΒΡΧαΗΏ≤Δ≤Μ °Ζ÷άμœκΓΘΫœΒΆΒΡ…ζΈοάϊ”ΟΕ»ΆυΆυΜαάΥΖ―ΫœΕύ“©ΈοΘ§ΒΦ÷¬≥…±ΨΧαΗΏΓΘΕχΡ…ΟΉΝΘΥδΡή¥οΒΫΫœΗΏΒΡ…ζΈοάϊ”ΟΕ»Θ§ΒΪΟφΝΌΉ≈≤ζ“ΒΜ·Ρ―Ε»¥σΓΔ≈ζΦδ≤ν“λœ‘÷χΒ»»±œίΘ§ΫœΡ― Βœ÷ΝΌ¥≤”Π”ΟΓΘ“Μ–©–¬ΦΦ θ ( άκΉ”»ή“ΚΓΔΈΔ’κ ) ΒєДϻϻΥΟ«Ω¥ΒΫΝΥΕύκΡ“©ΈοΩΎΖΰΗχ“©ΒΡ–¬œΘΆϊΓΘ
”…”ΎΕύκΡΒΡΈοάμΜ·―ß–‘÷ «ß≤νΆρ±πΘ§ΟΩ“Μ÷÷ΕύκΡ“©ΈοΕΦ”Π”–ΤδΉν ΚœΒΡΒίΥΆΖΫ ΫΓΘSNAC Ρή”––ßΒίΥΆΥς¬ξ¬≥κΡΒΪ»¥ΈόΖ® Βœ÷άϊά≠¬≥κΡΒΡΩΎΖΰΈϋ ’’β“Μ ΒάΐΧα–―Έ“Ο«Θ§’κΕ‘≤ΜΆ§–‘÷ ΒΡ“©Έο“Σ―Γ‘ώ≤ΜΆ§ΒΡΩΎΖΰΒίΥΆ≤Ώ¬‘ [11]ΓΘœ÷”–ΒΡ≤Ώ¬‘Ψυ”–Τδ“ΜΕ®ΒΡΨ÷œό–‘Θ§Μρ «ΈόΖ® Βœ÷…ζΈοάϊ”ΟΕ»ΒΡΗϋ¥σΧα…ΐΘ§Μρ «ΈόΖ®‘ΎΙΛ“’Υ°ΤΫ…œ Βœ÷ΝΩ≤ζΓΘ
ΥφΉ≈»ΥΟ«Ε‘ΕύκΡ“©Έο‘ΎΈΗ≥ΠΒά…ζάμΜΖΨ≥œ¬––ΈΣΒΡ…ν»κ―–ΨΩΦΑΒίΥΆ“©ΈοΦΦ θΒΡ≤ΜΕœΫχ≤ΫΘ§‘ΎΕύκΡ“©ΈοΩΎΖΰΒίΥΆΝλ”ρ÷–ΒΡ≥Δ ‘Μα‘Ϋά¥‘ΫΕύΘ§œύ–≈ΫΪά¥Μα”–ΗϋΕύΡήΧαΗΏΜΦ’ΏΥ≥”Π–‘ΒΡΩΎΖΰΕύκΡ≤ζΤΖΈ άΘ§ΈΣ¬ΐ–‘≤ΓΜΦ’Ώ¥χά¥ΗΘ“τΓΘ
Οβ‘π…υΟςΘΚ±ΨΈΡΈΣ––“ΒΫΜΝς―ßœΑΘ§Αφ»®Ιι‘≠Ής’ΏΦΑ‘≠‘”÷ΨΥυ”–Θ§»γ”–«÷»®Θ§Ω…ΝΣœΒ…Ψ≥ΐΓΘΈΡ’¬±ξΉΔ”–Ής’ΏΦΑΈΡ’¬≥ω¥ΠΘ§»γ–η‘ΡΕΝ‘≠ΈΡΦΑ≤ΈΩΦΈΡœΉΘ§Ω…‘ΡΕΝ‘≠‘”÷ΨΓΘ
