
摘 要 许多重要的生物过程的调节都通过蛋白⁃蛋白相互作用来实现的。一般,蛋白⁃蛋白作用的界面太大而不能被小分子药物选择性靶向,因此小分子药物很难高效特异性地阻断该类型的相互作用。此外,由于蛋白质药物很难透过细胞膜,它们也不能直接靶向细胞内的相互作用。由于当前药物分子的限制,发展下一代既能进入细胞膜又能特异性靶向蛋白⁃蛋白相互作用的分子成为新的研究热点。为了克服上述药物分子的缺点,Verdine 等发展了一种全碳支架的具有 α⁃螺旋结构的新型多肽,这种多肽被称作订书肽( stapledpeptides)。相比于天然多肽,订书肽有更高的酶解稳定性并且可以进入细胞膜,从而提高了它的药理性能。本文将从订书肽的化学合成、生物物理性能的表征和其在癌症和 HIV 治疗、信号通路的调节和肿瘤激活蛋白的抑制方面的生物应用详细介绍订书肽的最新进展。
蛋白⁃蛋白相互作用(PPI)在许多生物过程中扮演着重要的角色,例如病毒的自组装,细胞的增殖、生长、分化及程序性死亡。人类疾病中许多潜在的治疗靶标主要是蛋白⁃蛋白相互作用。由于大部分PPI 面比较大而且是不连续的平面,小分子试剂很难与其特异性紧密结合。目前,大约有 10% 的胞外疾病可以利用蛋白类药物来治疗。蛋白药物最大的缺点是不能透过细胞膜,因而无法靶向于胞内靶标。基于小分子和蛋白类药物的局限,发展能穿过细胞膜的多肽、蛋白质和核酸逐渐成为药物研究的前沿问题。最近生物大分子化学合成方面取得了一些新的进展, 为研发这类药物提供了有力的技术工具[1 ~ 21]。
2 订书肽的合成与修饰
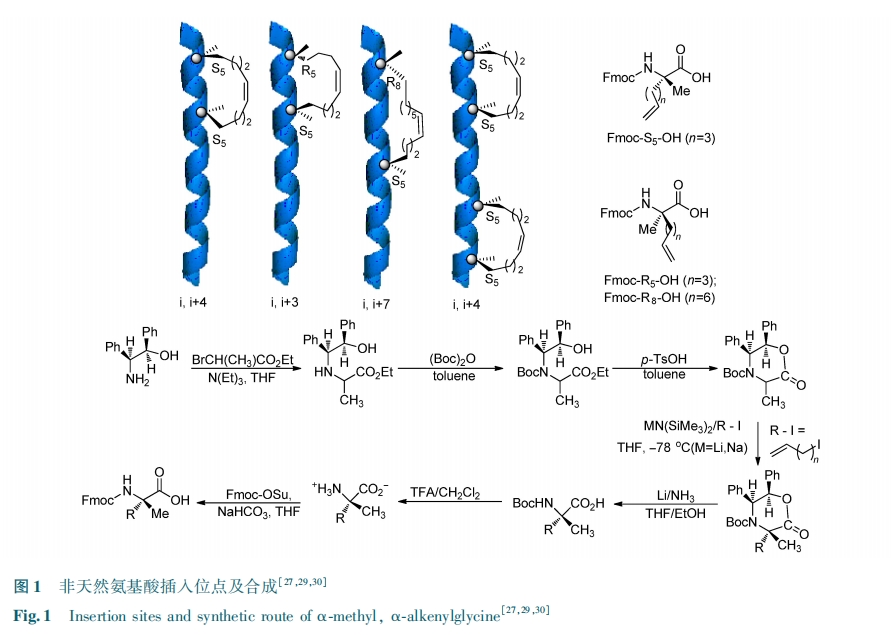
为了使订书肽与靶点蛋白质的相互作用不受外加非天然结构的影响,选择非天然氨基酸的插入位点至关重要。通过对蛋白质的核磁或晶体数据的分析,可以选择与靶向蛋白相互用的 α 螺旋多肽片段为研究对象,同时选择不参与靶向蛋白作用的氨基酸残基作为非天然氨基酸插入的潜在位点。如果缺乏上述数据,也可以首先合成一系列锁定多肽,然后通过活性筛选的方法选出最优结构的订书肽。通常,一对用于 RCM 反应的非天然氨基酸的合成流程[28,29]及插入在多肽序列的 i,i + 3 位,i,i + 4 位或者 i,i + 7 位的位点,如图 1。一般而言,选择合成的构象锁定 α 螺旋多肽的长度不超过 20 个氨基酸[30]。在设计多肽过程中,电荷也是影响订书肽功能的重要因素,正电荷有利于多肽跨膜,而负电荷不利于跨膜。研究发现,将正负电荷分别放在肽链的碳末端和氮末端可以产生额外的氢键结合,该结构能中和订书肽产生的大的偶极作用。
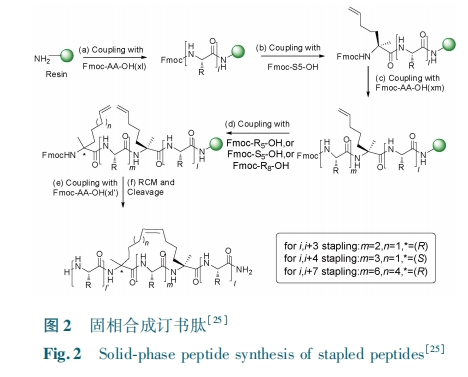
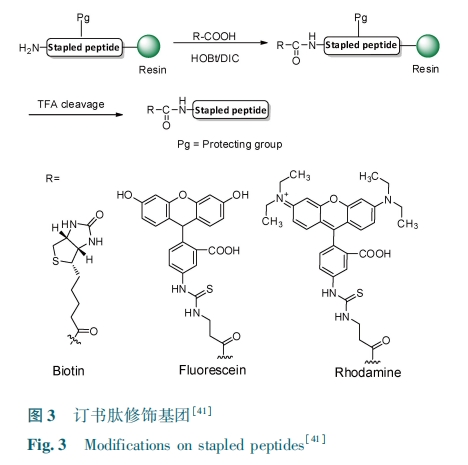
一般认为,由 30 个以上氨基酸残基组成的订书肽的合成不能通过固相合成一次高效合成,而需要多肽片段连接反应实现。2013 年, Pentelute 课题组, 用 “ 自 然 化 学 连 接 反 应 ” ( native chemicalligation,NCL) 的方法合成了长链非经典的订书肽(其侧链为半胱氨酸与六氟化苯或十氟联苯反应关环,而非 Verdine 型订书肽)从而使订书肽的得到更广泛的应用[19]。他们采用的连接方法为 Kent 发明的 NCL[1],它涉及氮端半胱氨酸肽段与碳端硫酯肽段。NCL 在蛋白质合成与半合成中取得了广泛的应用,但是碳端硫酯的 Fmoc 化学合成还存在挑战。近期的一个改进方法是我们发展的利用碳端酰肼代替硫酯进行多肽酰肼连接技术[6],该方法基本原理是在弱酸条件下多肽酰肼被亚硝酸氧化为酰基叠氮,并在芳基硫醇存在下原位转化为硫酯后与 N 端Cys 肽实现化学选择性连接反应。与硫酯相比,碳端酰肼肽可以直接采用 Fmoc 法固相合成高效得到,且易于自动化。此外,酰肼肽可以实现生物表达,使酰肼法应用范围更加广泛。
3 订书肽生物物理性质的表征
订书肽的结构和生物活性可以通过多种生物物理的实验进行表征,例如,圆二色光谱[32]、二维核磁共振谱、X 射线单晶衍射、荧光偏振和表面等离子体共振等。我们从以下 5 个方面来阐述该方面的进展。
核磁共振谱和 X 射线单晶衍射可以直接模拟出订书肽的实际结构,并可以确定订书肽与靶蛋白的结合位点,为进一步的订书肽的设计提供了结构基础。Phillips 等设计了靶向雌激素受体的订书肽[34]。通过对一系列锁定多肽的核磁与单晶衍射结构的表征,他们发现订书肽的碳碳支架会影响其与疏水蛋白表面的相互作用。
荧光偏振与表面等离子共振技术可以用于多肽与靶向蛋白结合能力的测定,并且能定量测量订书肽结合常数[35 ~ 38]。
偏振荧光的强弱程度与荧光分子的大小呈正相关,与其受激发时转动速度呈反相关。在固相多肽合成的过程中将一个荧光基团引入订书肽,如异硫氰酸荧光素。在溶液中,靶向蛋白与订书肽结合,可以产生荧光偏振,荧光偏振程度与多肽绑定在靶向蛋白的数量有关。通过测量偏振荧光,可以定量测定多肽与靶向蛋白的结合常数。另一种改进的荧光偏振方法可以测定不含荧光基团的订书肽阻断蛋白⁃蛋白相互作用的能力。在这种改进的荧光偏振中,含有荧光修饰的线性天然多肽与不含荧光修饰的订书肽混合在一起。通过对含有荧光标记的天然多肽荧光偏振降低程度的测量,来测量订书肽阻断天然多肽与蛋白靶向的结合能力。2009 年,Bautista等[39]用荧光偏振的方法,分析了构象锁定的 β 多肽与 hDM2 之间的结合能力。他们发现构象锁定的 β多肽与靶蛋白的结合能力与引入构象锁定支架的位置相关,有些位置可以明显增强结合能力,有些位置却会明显降低结合能力。
在表面等离子体共振实验中,首先将生物素修饰的订书肽固定在生物传感芯片上,然后将其暴露于靶向蛋白中。这样订书肽与靶向蛋白之间的相互作用可以被直接测定,并且可以得到结合的动力学过程。表面等离子体实验也可以用于测定订书肽抑制蛋白复合物形成的程度。将蛋白复合物固定在生物传感芯片上,然后将其暴露在订书肽中,这样可以测定订书肽对多蛋白复合物的结合与解离动力学影响。
从人工合成多肽到药物,多肽的酶解稳定性成为一个重要的指标,也是一个主要的障碍。由于蛋白水解酶水解酰胺键需要多肽有一个舒展的构象,因此用支架来锁定多肽的 α 螺旋结构可以有效地抑制多肽被蛋白水解酶水解。为了评估多肽的体外酶解稳定性,需要基于订书肽的序列选择蛋白水解酶。胰蛋白酶可以识别精氨酸 ( Arg) 和 赖 氨 酸(Lys),糜蛋白酶主要识别苯丙氨酸(Phe)、酪氨酸(Tyr)、亮氨酸(Leu)和甲硫氨酸(Met)。Bird 等[40]表征了 Bcl⁃2 域的订书肽,并进行了酶解稳定性的测定。他们通过对比荧光素标记的订书肽与荧光素标记的线性肽在胰蛋白酶存在下不同时间段的荧光强度变化来确定订书肽的酶解稳定性。对订书肽进行体内或体外的血清稳定性测定可以更加明确地表明多肽的酶解稳定性。
流式细胞计和共聚焦荧光显微镜这两种方法可以利用荧光标记订书肽来研究多肽的细胞通透性[41]。单个含有荧光分子的细胞可以便捷地被流式细胞计筛选出,这样可以精确了解订书肽的入膜效果。但是,流式细胞计数不能观察订书肽在细胞中的具体位置。由于黏附在细胞表面的订书肽会产生入膜的错误信息,在分析订书肽入膜性时,必须用胰蛋白酶除去吸附细胞表面的订书肽。共聚焦荧光显微镜法研究的细胞数量明显要少于流式细胞计所研究的数目。需要指出的是,共聚焦荧光显微镜法可以研究订书肽的入膜的更多细节,如订书肽在细胞内的具体位置,但是共聚焦显微镜法只能定性的判断多肽是否能进入细胞。最近发展了一种可以定量测定多肽入膜性的荧光显微镜法,也可以直接定量比较多肽的入膜性差异,但不能提供可视化信息。
Muppidi 等[31]采用流式细胞计和共聚焦荧光显微镜研究了精氨酸修饰的订书肽的入膜过程。通过流式细胞计的方法,他们发现精氨酸修饰的订书肽可以明显提高多肽的入膜性;利用共聚焦显微镜法,他们进一步的确认了该结果,并提出精氨酸修饰的订书肽的入膜可能是通过内吞机理实现的。
理论计算结构表明订书肽中支架的局部稳定作用比 α 螺旋程度对生物活性的影响可能更大,测定订书肽的生物活性也成为必不可少的一步。体外免疫共沉淀可以测定订书肽在细胞环境下与靶蛋白的结合能力[42 ~ 45]。订书肽的免疫共沉淀过程可以简述为用抗体将目标蛋白特异性沉淀下来,同时与目标蛋白结合的订书肽也一起被沉淀。常用两种订书肽进行该实验,一种是利用荧光素标记的订书肽,另一种是利用生物素标记的订书肽。具体步骤主要为,在细胞裂解液中加入靶蛋白的特异性抗体,孵育一段时间后,加入与抗体特异性结合的琼脂糖上的蛋白 A 或蛋白 G。当订书肽与靶蛋白结合时,订书肽也同时被沉降;沉淀复合物通过变性聚丙烯酰胺凝胶电泳又被分开;最后采用免疫印迹实验进一步证实构象锁定肽与靶蛋白结合。
订书肽是否具有潜在的医学应用是通过活体内其功能的测量来评估的。将订书肽注入到活的患病动物体内,观察和测定动物体内各项生理指标以确定订书肽的生物活性。
4 订书肽的生物医学功能与应用
到目前为止,国际上有许多课题组都在从事与订书肽相关的研究,并取得了一定的成果。订书肽在治疗癌症、抑制艾滋病和丙型肝炎以及调节信号通路等方面的应用均有报道。下面主要将从这几个方面简单介绍一下订书肽的医学应用。
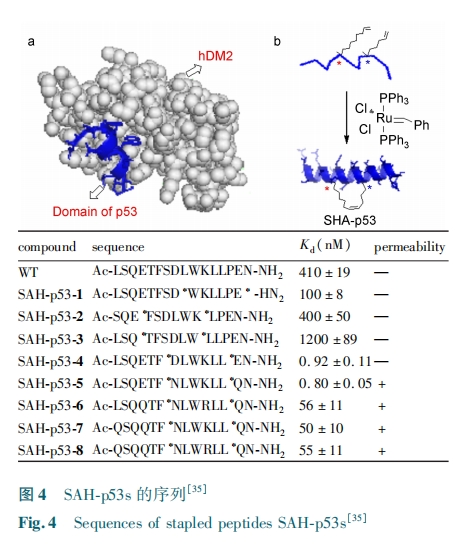
2010 年, Bernal 等[46] 对 SAH⁃p53 8 调控 p53进行 了 进 一 步 的 研 究。hDMx 的 结 构 分 析 发 现hDMx 抑制 p53 的反式激活的 α 螺旋与 hDM2 的模式极为相似。因而,他们测试 SAH⁃p53 8 对 hDMx的靶向抑制作用,并且研究了 hDMx 被抑制后的体内 p53 功能变化,实验结果表明,SAH⁃p53 8 可以有效地抑制 hDMx。SAH⁃p53 8 既可以靶向 hDM2 也可以靶向 hDMx,并且 SAH⁃p53 8 对于 hDMx 靶向能力比对 hDM2 的靶向能力高 25 倍。SAH⁃p53 8 靶向细胞里的 hDMx,阻断 p53⁃hDMX 复合物的形成,从而激活 p53 的肿瘤抑制通路。因此,对 hDM2 和hDMx 双靶向的抑制剂可以更好地用于治疗癌症。

Bax 是 Bcl⁃2 家族蛋白中一个促凋亡因子。它位于细胞基质中,其被激活后会诱导细胞的死亡。Bcl⁃2 等抗凋亡蛋白可以与 Bax 相互作用,抑制细胞的死亡。目前,科学家一致认为细胞的凋亡是通过细胞凋亡蛋白与抗凋亡蛋白相互作用来调节的,然而在凋亡应激反应中激活 Bax 和 Bak 的机理仍然存在争论。Gavathiotis 等发现订书肽 SAHBs 可以直接激活 Bax 调节的线粒体凋亡[42]。通过 Bim SAHB与 Bax 复合物的二维15N⁃1H 异核单量子相关核磁共振谱与顺磁弛豫增强核磁共振实验,他们确定 Bax的激活位点与抗凋亡蛋白的典型模式完全不同。Bax 激活反应引起了一系列动态连续变化,包括构象的改变与齐聚反应。通过一系列订书肽与 BAax结合实验发现,只要碳链支架不是位于结合位点,Bim SAHB 均能有效地与 Bax 作用。该工作为癌症的治疗提供了一条新的思路,即建立细胞凋亡调节的一个新的靶标,寻找订书肽来激活癌细胞中的细胞凋亡通路。
人类免疫缺陷病毒(HIV)属于反转录病毒的一种。HIV 通过破坏人体的免疫能力,导致免疫系统对抗原失去抵抗力,从而引发各种疾病包括癌症。HIV⁃1 衣壳蛋白在病毒组装中扮演着重要的角色,成为新型艾滋病疗法的一个重要的靶标。研究者曾报道一个含 12 个氨基酸的带有 α 螺旋结构的多肽(CAI) 可以在体外靶向衣壳的碳端区域( C⁃CA)阻断病毒衣壳的组装[51]。但是,该多肽因不能进入细胞膜而在生物体内不能发生作用,不适合作为抗病毒的药物。CAI 在复合物 CAI⁃ C⁃CA 结合的结构已经通过高分辨 X 射线晶体衍射法被解析[52]。Zhang 等[38] 通过结构优化将 CAI 转变成可以进入细胞膜的订书肽(NYAD⁃1)。首先,他们利用圆二色谱测试溶液中 NYAD⁃1 与 CAI 的二级结构,发现CAI 主要以无规则卷曲的形式存在,并无典型的 α螺旋结构。这一结果间接支持了结合诱导 CAI 的构象发生变化导致 CAI 与 C⁃CA 复合物的形成。不同的是,NYAD⁃1 有着明显的 α 螺旋结构,其螺旋程度大约 为 80% 。通 过 核 磁 谱 图 映 射, 他 们 发 现NYAD⁃1 和 CAI 分别与 C⁃CA 结合的化学位移非常相似,从而证明其结合方式也非常相似。通过共聚焦显微镜实验,证实订书肽可以进入细胞膜;通过体外细胞实验,他们证实了 NYAD⁃1 可以抑制病毒的组装。在病毒侵染细胞实验中,NYAD⁃1 没有任何活性,表明订书肽对病毒进入细胞膜的过程没有抑制作用。这也进一步确定 NYAD⁃1 通过直接结合在CA 上影响二聚界面的形成,阻碍成熟的和未成熟的病毒 衣 壳 的 形 成, 减 少 病 毒 感 染 人 体 内 细 胞。NYAD⁃1 是对于抗 HIV⁃1 有着广谱的抗病毒活性,有着被优化成为一种新的治疗艾滋病药物的潜力。
许多链段长的多肽由于结构的丧失和酶解稳定性差而致使其生物利用性低。恩弗韦肽是由 36 个氨基酸组成的多肽,是第一个可以阻断 HIV⁃1 进入人体的膜融合抑制剂。它通过靶向病毒的融合片段来抑制 HIV⁃1 病毒的感染。但是,由于生物稳定性差,不能口服,恩弗韦肽一直被限制成为补救型的医疗选择。为了克服用作医疗多肽的酶解稳定性差,Bird 等[53]设计并合成了碳碳骨架的构象锁定的恩弗韦肽(如表 3 所示),并对其进行了生物物理和生物药理学影响研究。他们选取恩弗韦肽(i,i + 4)的位置插入非天然氨基酸,经过烯烃复分解形成构象锁定的恩弗韦肽。根据 gp41 的晶体结构选择非天然氨基酸插入位点以确保订书肽不会打断 HR1 与HR2 直接相互作用的严格的疏水界面。经过糜蛋白酶的酶解稳定性测试,他们发现含有一个支架的恩弗韦肽的酶解稳定远强于未修饰的恩弗韦肽;含有两个支架的恩弗韦肽比一个支架的恩弗韦肽有着更强的酶解稳定性。抗毒活性实验表明含有两个支架的恩弗韦肽可以有效地抑制病毒的感染。此外,小鼠体内酶解稳定性实验发现具有两个支架的订书肽在体内的半衰期有着显著地提高,弥补了多肽类HIV⁃1 膜融合抑制剂的药代动力学缺陷。经过小鼠实验发现打两个支架的恩弗韦肽有一定的口服利用度,在灵长类动物中其潜在的口服利用度有待进一步的研究。

Notch 信号通路是由局部细胞间相互作用而产生的,并与肿瘤形成和某些神经系统疾病有着密切的关系,具有复杂多样的功能,如参与造血、T 细胞发育和血管生成等生理过程。Notch 蛋白参与一个保守的信号通路,可以调节细胞的分化、增殖和死亡。Notch 信号的持续时间跟强度被严格调控,在许多疾病中 Notch 蛋白功能丧失的突变已经被发现。Notch 功能获得性的突变与癌症的发生有关。在急性 T 淋巴细胞白血病表达的 MAML1 的显性抑制碎片( dnMAML1) 表明有着抑制 Notch 信号和细胞增殖的能力。一个构象锁定的 dnMAML1 碎片,可能抑制全长的 MAML1 与 ICN1⁃CSL 复合物的结合。区别于未修饰的多肽,订书肽有着更高的结合靶向蛋白能力,更高的代谢稳定性和血浆半衰期。
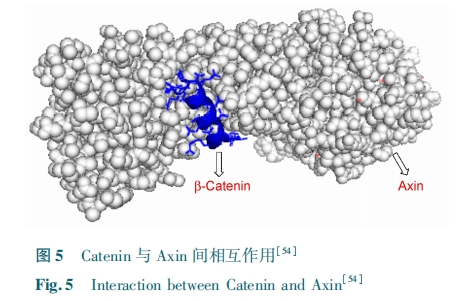
最近,Cui 等[54]设计了可以激活 Wnt 信号通路的能入膜的订书肽。该多肽旨在打破 Axin 与 β⁃连环蛋白之间的作用。他们化学合成了两条订书肽SAHPA1 与 SAHPA2;通过圆二色谱实验发现,与线性的多肽 Axin 相比,两条合成的订书肽的 α 螺旋程度明 显 提 高。体 外 pull⁃down 实 验 数 据 表 明SAHPA1 结合 β 连环蛋白的结合能力比 SAHPA2 更强。细 胞 入 膜 性 实 验 和 免 疫 共 沉 淀 实 验 表 明SAHPA1 通过与 β⁃连环蛋白的结合可以有效地打破内源的 Axin⁃β⁃连环蛋白复合物的生成。他们探索了对转录活性的影响,表明 SAHPA1 可以高度特异性地激活 Wnt 信号。
5 订书肽的前景与展望
Verdine 等首次发明出碳碳链支架的订书肽至今,从合成方法和生物应用两方面,均得到了快速的发展。传统小分子体积过小不能有效地调节蛋白蛋白相互作用。订书肽不仅可以作为一个潜在的医疗靶向,而且可以作为一个新的工具用于识别新的反应位点及新的配体等。订书肽比小分子有更多的选择性,其价格又远低于抗体等生物制剂,而且可以入膜。同样具有稳定空间折叠结构还有小蛋白(尤其是环蛋白)[56 ~ 65],这些分子的独特性质,使得它们成为未来药物发展值得探索的一个方向。随着多肽连接技术的发展,大型订书肽的合成成为现实。这将为发展新一代蛋白类药物提供强有力的工具。我们有理由相信在未来几年订书肽类的药物以及小蛋白药物会不断地投入到临床,并获得长足的发展。
免责声明:本文为行业交流学习,版权归原作者及原杂志所有,如有侵权,可联系删除。文章标注有作者及文章出处,如需阅读原文及参考文献,可阅读原杂志。

电话:0551-65177703 邮箱:pb@peptidesbank.com 地址:安徽省合肥市四川路868号云谷创新园A6栋3层
合肥肽库生物(Taikubio)只为有资质的科研机构、医药企业基于科学研究或药证申报的用途提供医药研发服务, 不为任何个人或者非科研性质的、非用于药证申报使用等其他用途提供服务。