
’Σ “Σ –μΕύ÷Ί“ΣΒΡ…ζΈοΙΐ≥ΧΒΡΒςΫΎΕΦΆ®ΙΐΒΑΑΉ⁃ΒΑΑΉœύΜΞΉς”Οά¥ Βœ÷ΒΡΓΘ“ΜΑψΘ§ΒΑΑΉ⁃ΒΑΑΉΉς”ΟΒΡΫγΟφΧΪ¥σΕχ≤ΜΡή±Μ–ΓΖ÷Ή”“©Έο―Γ‘ώ–‘Α–œρΘ§“ρ¥Υ–ΓΖ÷Ή”“©ΈοΚήΡ―ΗΏ–ßΧΊ“λ–‘ΒΊΉηΕœΗΟάύ–ΆΒΡœύΜΞΉς”ΟΓΘ¥ΥΆβΘ§”…”ΎΒΑΑΉ÷ “©ΈοΚήΡ―ΆΗΙΐœΗΑϊΡΛΘ§ΥϋΟ«“≤≤ΜΡή÷±Ϋ”Α–œρœΗΑϊΡΎΒΡœύΜΞΉς”ΟΓΘ”…”ΎΒ±«Α“©ΈοΖ÷Ή”ΒΡœό÷ΤΘ§ΖΔ’Ιœ¬“Μ¥ζΦ»ΡήΫχ»κœΗΑϊΡΛ”÷ΡήΧΊ“λ–‘Α–œρΒΑΑΉ⁃ΒΑΑΉœύΜΞΉς”ΟΒΡΖ÷Ή”≥…ΈΣ–¬ΒΡ―–ΨΩ»»ΒψΓΘΈΣΝΥΩΥΖΰ…œ ω“©ΈοΖ÷Ή”ΒΡ»±ΒψΘ§Θ÷ΘεΘρΘδΘιΘνΘε Β»ΖΔ’ΙΝΥ“Μ÷÷»ΪΧΦ÷ßΦήΒΡΨΏ”– ΠΝ⁃¬ί–ΐΫαΙΙΒΡ–¬–ΆΕύκΡΘ§’β÷÷ΕύκΡ±Μ≥ΤΉςΕ© ικΡΘ® ΘσΘτΘαΘπΘλΘεΘδΘπΘεΘπΘτΘιΘδΘεΘσΘ©ΓΘœύ±»”ΎΧλ»ΜΕύκΡΘ§Ε© ικΡ”–ΗϋΗΏΒΡΟΗΫβΈ»Ε®–‘≤Δ«“Ω…“‘Ϋχ»κœΗΑϊΡΛΘ§¥”ΕχΧαΗΏΝΥΥϋΒΡ“©άμ–‘ΡήΓΘ±ΨΈΡΫΪ¥”Ε© ικΡΒΡΜ·―ßΚœ≥…ΓΔ…ζΈοΈοάμ–‘ΡήΒΡ±μ’ςΚΆΤδ‘ΎΑ©÷ΔΚΆ Θ»Θ…Θ÷ ÷ΈΝΤΓΔ–≈Κ≈Ά®¬ΖΒΡΒςΫΎΚΆ÷ΉΝωΦΛΜνΒΑΑΉΒΡ“÷÷ΤΖΫΟφΒΡ…ζΈο”Π”ΟœξœΗΫι…ήΕ© ικΡΒΡΉν–¬Ϋχ’ΙΓΘ
ΒΑΑΉ⁃ΒΑΑΉœύΜΞΉς”ΟΘ®Θ–Θ–Θ…Θ©‘Ύ–μΕύ…ζΈοΙΐ≥Χ÷–Αγ―ίΉ≈÷Ί“ΣΒΡΫ«…ΪΘ§άΐ»γ≤ΓΕΨΒΡΉ‘ΉιΉΑΘ§œΗΑϊΒΡ‘ω÷≥ΓΔ…ζ≥ΛΓΔΖ÷Μ·ΦΑ≥Χ–ρ–‘ΥάΆωΓΘ»ΥάύΦ≤≤Γ÷––μΕύ«±‘ΎΒΡ÷ΈΝΤΑ–±ξ÷ς“Σ «ΒΑΑΉ⁃ΒΑΑΉœύΜΞΉς”ΟΓΘ”…”Ύ¥σ≤ΩΖ÷Θ–Θ–Θ… Οφ±»Ϋœ¥σΕχ«“ «≤ΜΝ§–χΒΡΤΫΟφΘ§–ΓΖ÷Ή” ‘ΦΝΚήΡ―”κΤδΧΊ“λ–‘ΫτΟήΫαΚœΓΘΡΩ«ΑΘ§¥σ‘Φ”– Θ±ΘΑΘΞ ΒΡΑϊΆβΦ≤≤ΓΩ…“‘άϊ”ΟΒΑΑΉάύ“©Έοά¥÷ΈΝΤΓΘΒΑΑΉ“©ΈοΉν¥σΒΡ»±Βψ «≤ΜΡήΆΗΙΐœΗΑϊΡΛΘ§“ρΕχΈόΖ®Α–œρ”ΎΑϊΡΎΑ–±ξΓΘΜυ”Ύ–ΓΖ÷Ή”ΚΆΒΑΑΉάύ“©ΈοΒΡΨ÷œόΘ§ΖΔ’ΙΡή¥©ΙΐœΗΑϊΡΛΒΡΕύκΡΓΔΒΑΑΉ÷ ΚΆΚΥΥα÷πΫΞ≥…ΈΣ“©Έο―–ΨΩΒΡ«Α―ΊΈ ΧβΓΘΉνΫϋ…ζΈο¥σΖ÷Ή”Μ·―ßΚœ≥…ΖΫΟφ»ΓΒΟΝΥ“Μ–©–¬ΒΡΫχ’ΙΘ§ ΈΣ―–ΖΔ’βάύ“©ΈοΧαΙ©ΝΥ”–ΝΠΒΡΦΦ θΙΛΨΏΘέΘ± ΓΪ Θ≤Θ±ΘίΓΘ
ΉνΫϋ―–ΨΩ±μΟςΘ§ΨΏ”– ΠΝ ¬ί–ΐΫαΙΙΚΆΗΜΚ§’ΐΒγΚ…ΒΡΕύκΡΘέΘ≤Θ≤ ΓΪ Θ≤Θ¥ΘίΩ…“‘¥©ΙΐœΗΑϊΡΛΓΘ“ρ¥ΥΘ§‘Ϋά¥‘ΫΕύΒΡ―–ΨΩΩΣ ΦΙΊΉΔΚ§”– ΠΝ ¬ί–ΐΫαΙΙΒΡΕύκΡΒΡΚœ≥…”κ”Π”ΟΓΘ»ΜΕχΘ§ΕύκΡ“ΜΒ©¥”ΡΗΧε…œΖ÷άκΨΆ≤ΜΡή±Θ≥÷‘≠”–ΒΡΕΰΦΕΫαΙΙΘ§”…”ΎΙΙœσΒΡ≤ΜΈ»Ε®Θ§ΕύκΡ”κΉς”ΟΒΑΑΉΒΡΫαΚœΡήΝΠΖ«≥Θ»θΘ§ΕχΤ’Ά®ΒΡœΏ–‘ΕύκΡ≤ΜΡήΆΗΙΐœΗΑϊΡΛΘ§«““Ή±ΜΒΑΑΉΟΗΥ°ΫβΘέΘ≤ΘΒΘίΓΘΜυ”Ύ¥ΥΘ§»ΥΟ«≤ΜΕœ≥Δ ‘ΖΔ’ΙΈ»Ε® ΠΝ ¬ί–ΐΫαΙΙΒΡΖΫΖ®Θ§άΐ»γΘ§άϊ”ΟΕΰΝρΦϋΜρΖ÷Ή”ΡΎθΘΑΖΦϋΉςΈΣ÷ßΦήΓΘ»ΜΕχΘ§’β–©÷ßΦή‘Ύ…ζάμΜΖΨ≥œ¬Ψυ≤ΜΡήΈ»Ε®¥φ‘ΎΓΘΘ≤ΘΑΘΑΘΑ ΡξΘ§Θ÷ΘεΘρΘδΘιΘνΘε Β»ΖΔ’ΙΝΥ“Μ÷÷”ΟΧΦΧΦΦϋΉςΈΣ÷ßΦήά¥Έ»Ε®ΕύκΡ ΠΝ ¬ί–ΐΫαΙΙΒΡΖΫΖ®ΘέΘ≤ΘΕΘίΓΘ”… ΗΟ ΖΫ Ζ® ΒΟ ΒΫ ΒΡ Εύ κΡ ≥Τ ΈΣ Ε© ι κΡ Θ® ΘσΘτΘαΘπΘλΘεΘδΘπΘεΘπΘτΘιΘδΘεΘσΘ©ΓΘΕ© ικΡ”–Ή≈ΗϋΗΏΒΡ ΠΝ ¬ί–ΐ≥ΧΕ»Θ§”κΑ–±ξΒΡΫαΚœΡήΝΠ‘ωΦ” ΘΒ ΓΪ ΘΒ ΘΑΘΑΘΑ ±ΕΓΘ¥ΥΆβΘ§Ε© ικΡΡήΆΗΙΐœΗΑϊΡΛΘ§Ρ―±ΜΒΑΑΉΟΗΥ°ΫβΘ§‘Ύ…ζΈοΧεΡΎΒΡΑκΥΞΤΎΫœ≥ΛΓΘ±ΨΈΡΫΪ÷ς“Σ¥”Κœ≥…ΖΫΖ®ΓΔάμΜ·–‘÷ ΚΆ…ζΈο―ßΙΠΡήΦΑ«ΑΨΑ’ΙΆϊά¥ Θ¥ ΗωΖΫΟφΫι…ήΕ© ικΡΓΘ±μ Θ± ΈΣ»ΐ÷÷“©ΈοΖ÷Ή””≈»±Βψ±»ΫœΓΘ
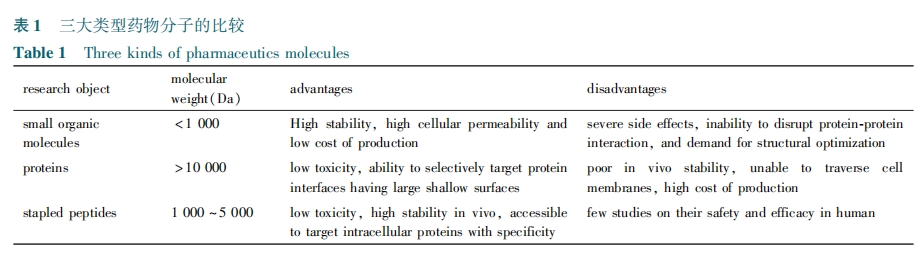
Θ≤ Ε© ικΡΒΡΚœ≥…”κ–ό Έ
Ε© ικΡΒΡΚœ≥…≤Ώ¬‘»γœ¬Θ§ Ήœ»‘ΎΙΧœύΚœ≥…ΕύκΡΝ¥Ιΐ≥Χ÷–“ΐ»κΝΫΗω ΠΝ⁃ΦΉΜυΘ§ΠΝ⁃œ©ΜυΖ«Χλ»ΜΑ±ΜυΥαΘ§»ΜΚσΆ®Ιΐœ©ΧΰΗ¥Ζ÷ΫβΖ¥”ΠΘ®Θ“ΘΟΘΆΘ© ΜΖΜ·Θ§ΒΟΒΫΕ© ικΡΘέΘ≤ΘΖΘίΓΘ
ΠΝ⁃ΦΉΜυΘ§ΠΝ⁃œ©ΜυΖ«Χλ»ΜΑ±ΜυΥα“ρΚ§”– ÷–‘÷––ΡΘ§–η≤…”Ο ÷–‘ΜυΆ≈”’ΒΦά¥Κœ≥…ΓΘ“‘ Θ”⁃–Ά ΠΝ⁃ΦΉΜυΘ§ΠΝ⁃œ©ΜυΖ«Χλ»ΜΑ±ΜυΥαΒΡΚœ≥…ΈΣάΐΘ§Έ“Ο«ΦρΒΞΫι…ήΚœ≥…ΗΟάύΈο÷ ΒΡ“ΜΑψΖΫΖ®Θ§ΦϊΆΦ Θ±ΓΘ Ήœ»Θ§‘Ύ»ΐ““ΑΖ¥ΏΜ·œ¬Θ§ ÷–‘Η®Μυ ‘ΦΝΘ®Θ±Θ“Θ§Θ≤Θ”Θ©⁃Θ≤⁃ΑΖΜυ⁃Θ±Θ§Θ≤⁃Εΰ±Ϋ““¥Φ”κΘ≤⁃δε±ϊΥα““θΞΖΔ…ζ«ΉΚΥ»Γ¥ζΖ¥”ΠΘ§≤Δ≤…”ΟΘ®Θ¬ΘοΘψΘ©Θ≤Θœ±ΘΜΛΉ‘”…ΑΖΜυΘΜΫ”œ¬ά¥Θ§‘ΎΕ‘ΦΉΜυ±ΫΜ«ΥαΒΡ¥ΏΜ·œ¬Θ§ΜΖΉ¥ΒΡΚ§”– ÷–‘Η®ΜυΒΡ±ϊΑ±Υα«ΑΧεΒΟ“‘ΗΏ–ß…ζ≥…Θ§‘ΎΦν–‘¥ΏΜ·ΦΝΚΆΒΆΈ¬œ¬Θ§œ©ΜυΜυΆ≈ΗΏ–ßΝΔΧε―Γ‘ώ–‘ΒΊΝ§Ϋ”ΒΫΑ±ΜυΥαΒΡ ΠΝ⁃ΈΜΓΘΉνΚσΘ§‘ΎΑ±Μυο°ΧθΦΰœ¬Ά―»Ξ ÷–‘Η®ΜυΘ§≤Δ≤…”Ο ΘΤΘμΘοΘψ ΜυΆ≈±ΘΜΛ ΠΝ⁃ΑΖΜυΓΘ
Θ≤.Θ± Ζ«Χλ»ΜΑ±ΜυΥα≤ε»κΈΜΒψΒΡ―Γ‘ώ
ΈΣΝΥ ΙΕ© ικΡ”κΑ–ΒψΒΑΑΉ÷ ΒΡœύΜΞΉς”Ο≤Μ ήΆβΦ”Ζ«Χλ»ΜΫαΙΙΒΡ”ΑœλΘ§―Γ‘ώΖ«Χλ»ΜΑ±ΜυΥαΒΡ≤ε»κΈΜΒψ÷ΝΙΊ÷Ί“ΣΓΘΆ®ΙΐΕ‘ΒΑΑΉ÷ ΒΡΚΥ¥≈ΜρΨßΧε ΐΨίΒΡΖ÷ΈωΘ§Ω…“‘―Γ‘ώ”κΑ–œρΒΑΑΉœύΜΞ”ΟΒΡ ΠΝ ¬ί–ΐΕύκΡΤ§ΕΈΈΣ―–ΨΩΕ‘œσΘ§Ά§ ±―Γ‘ώ≤Μ≤Έ”κΑ–œρΒΑΑΉΉς”ΟΒΡΑ±ΜυΥα≤–ΜυΉςΈΣΖ«Χλ»ΜΑ±ΜυΥα≤ε»κΒΡ«±‘ΎΈΜΒψΓΘ»γΙϊ»±ΖΠ…œ ω ΐΨίΘ§“≤Ω…“‘ Ήœ»Κœ≥…“ΜœΒΝ–ΥχΕ®ΕύκΡΘ§»ΜΚσΆ®ΙΐΜν–‘…Η―ΓΒΡΖΫΖ®―Γ≥ωΉν”≈ΫαΙΙΒΡΕ© ικΡΓΘΆ®≥ΘΘ§“ΜΕ‘”Ο”Ύ Θ“ΘΟΘΆ Ζ¥”ΠΒΡΖ«Χλ»ΜΑ±ΜυΥαΒΡΚœ≥…Νς≥ΧΘέΘ≤ΘΗΘ§Θ≤ΘΙΘίΦΑ≤ε»κ‘ΎΕύκΡ–ρΝ–ΒΡ ΘιΘ§Θι ΘΪ Θ≥ ΈΜΘ§ΘιΘ§Θι ΘΪ Θ¥ ΈΜΜρ’Ώ ΘιΘ§Θι ΘΪ ΘΖ ΈΜΒΡΈΜΒψΘ§»γΆΦ Θ±ΓΘ“ΜΑψΕχ―‘Θ§―Γ‘ώΚœ≥…ΒΡΙΙœσΥχΕ® ΠΝ ¬ί–ΐΕύκΡΒΡ≥ΛΕ»≤Μ≥§Ιΐ Θ≤ΘΑ ΗωΑ±ΜυΥαΘέΘ≥ΘΑΘίΓΘ‘Ύ…ηΦΤΕύκΡΙΐ≥Χ÷–Θ§ΒγΚ…“≤ «”ΑœλΕ© ικΡΙΠΡήΒΡ÷Ί“Σ“ρΥΊΘ§’ΐΒγΚ…”–άϊ”ΎΕύκΡΩγΡΛΘ§ΕχΗΚΒγΚ…≤Μάϊ”ΎΩγΡΛΓΘ―–ΨΩΖΔœ÷Θ§ΫΪ’ΐΗΚΒγΚ…Ζ÷±πΖ≈‘ΎκΡΝ¥ΒΡΧΦΡ©ΕΥΚΆΒΣΡ©ΕΥΩ…“‘≤ζ…ζΕνΆβΒΡ«βΦϋΫαΚœΘ§ΗΟΫαΙΙΡή÷–ΚΆΕ© ικΡ≤ζ…ζΒΡ¥σΒΡ≈ΦΦΪΉς”ΟΓΘ
Θ≤. Θ≤ Ε© ικΡΒΡΚœ≥…
Ά®≥Θ≤…”Ο ΘΤΘμΘοΘψ⁃ΙΧœύΕύκΡΚœ≥…ΖΫΖ®÷Τ±ΗΕ© ικΡΓΘΤ’Ά®Α±ΜυΥαΒΡ≤ύΝ¥”…Ε‘Υα≤ΜΈ»Ε®ΒΡ±ΘΜΛΜυ±ΘΜΛΘ§ΠΝ⁃Α±Μυ”…±ΜΕ‘Φν≤ΜΈ»Ε®ΒΡ ΘΤΘμΘοΘψ ±ΘΜΛΜυ±ΘΜΛΓΘΝΫΗω”Ο”ΎΙΙœσΥχΕ®ΒΡΑ±ΜυΥαΨυΈΣ ΠΝ⁃ΦΉΜυΘ§ΠΝ⁃œ©ΜυΖ«Χλ»ΜΑ±ΜυΥαΓΘ’βΝΫΗωΖ«Χλ»ΜΑ±ΜυΥαΦδΗτ“ΜΑψΈΣΝΫΗωΓΔ»ΐΗωΜρ’ΏΝυΗωΑ±ΜυΥαΘ§Τδ÷– ΘιΘ§ Θι ΘΪ Θ≥⁃ΈΜΚΆ ΘιΘ§ Θι ΘΪ Θ¥⁃ΈΜΈ»Ε®“ΜΗω ΠΝ⁃¬ί–ΐΘ§ΘιΘ§ Θι ΘΪ ΘΖ⁃ΈΜΈ»Ε®ΝΫΗω ΠΝ⁃¬ί–ΐΘ§»γΆΦ Θ±ΓΘΩ…“‘ΗυΨίΧΦΕΥΙΌΡήΆ≈ά¥―Γ‘ώ ς÷§Θ§»γ–ηΧΦΕΥ±ΘΝττ»ΜυΘ§Ω…“‘―Γ‘ώ Θ≤⁃ΘψΘλ⁃ΘτΘρΘτ ς÷§Μρ ΘΉΘαΘνΘγ ς÷§ΘΜ»γ–ηΧΦΕΥΈΣθΘΑΖΘ§Ω…“‘―Γ‘ώ Θ“ΘιΘνΘκ ΘΝΘμΘιΘδΘε⁃ΘΝΘΆ ς÷§ΓΘΕύκΡΚœ≥…Άξ≥…ΚσΘ§≤…”Ον…ΉςΈΣ¥ΏΜ·ΦΝΫχ––œ©ΧΰΗ¥Ζ÷ΫβΖ¥”ΠΙΊΜΖΘ§–Έ≥…ΧΦΧΦΦϋ÷ßΦήΓΘΗυΨίΚσ–χ Β―ι–η“ΣΘ§Ε© ικΡΩ…“‘‘ΎΙΧœύ…œΫχ––Ϋχ“Μ≤ΫΒΡ–ό ΈΓΘΉνΚσΘ§ΫΪΡΩ±ξΕύκΡ¥” ς÷§…œ«–Ηνœ¬ά¥Ϋχ––¥ΩΜ·Θ§ΆΦ Θ≤ ΈΣ ΘιΘ§ Θι ΘΪ Θ¥⁃ΈΜΕ© ικΡΚœ≥…ΒΡΝς≥Χ Ψ“βΆΦΓΘ
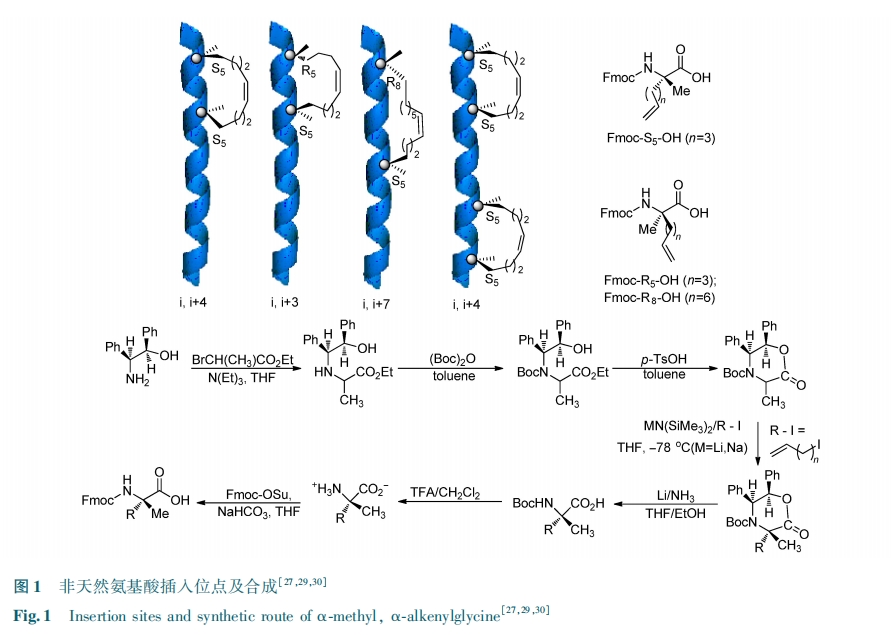
Θ≤.Θ≥ Ε© ικΡΒΡ–ό Έ
‘Ύ¥” ς÷§…œ«–Ηνœ¬Ε© ικΡ÷°«ΑΘ§ΫΪΕύκΡΒΣΕΥ»Ξ±ΘΜΛΘ§¬ψ¬Ε≥ω ΠΝ⁃Α±ΜυΘ§ΦΧΕχΩ…“‘‘ΎΗΟΈΜ÷ΟΫχ––≤ΜΆ§ΒΡ–ό ΈΓΘ»γΙϊ≤Μ–η“ΣΑ±ΜυΒΡΒγΚ…Θ§Ω…“‘Ϋχ––““θΘΜ·–ό ΈΘΜ»γΙϊ–η“ΣΤδΥϊ–ό ΈΘ§Ω…“‘Ϋχ“Μ≤ΫΫχ–––ό ΈΓΘΕ© ικΡΒΡ–ό Έ¥σ÷¬Ω…“‘Ζ÷ΈΣΝΫάύΘΚ”ΪΙβΥΊάύ±ξ«©Θ®÷ς“Σ «”ΪΙβΥΊΘ©ΚΆ«ΉΚΆάύ±ξ«©Θ®÷ς“Σ «…ζΈοΥΊΘ©ΓΘΆΦ Θ≥ΈΣ÷ς“ΣΒΡΕ© ικΡΒΡ–ό ΈΙΐ≥ΧΓΘΕ© ικΡΒΣΕΥ–ό Έ”ΪΙβΥΊΩ…“‘”Οά¥―–ΨΩΕύκΡΒΡœΗΑϊΈϋ ’ΚΆ…ζΈοΈοάμ–‘÷ ΘΜΒΣΕΥ–ό Έ…ζΈοΥΊΩ…”Οά¥―–ΨΩΕύκΡΒΡ…ζΈοΈοάμ–‘÷ ΚΆΤάΦέ…ζΈοΧεΡΎΒΡΑ–œρΖ¥”ΠΓΘΒ±–ό ΈΒΣΕΥΒΡ ±ΚρΘ§±Ί–κΩΦ¬«–ό ΈΒΡΈΜ÷Ο «ΖώΜα”ΑœλΕ© ικΡ”κΑ–œρΒΑΑΉΒΡΫαΚœΡήΝΠΓΘ¥ΥΆβΘ§“≤Ω…“‘Ε‘Ε© ικΡΒΡΧΦΕΥΫχ–––ό ΈΓΘΘ≤ΘΑΘ±Θ±ΡξΘ§ΘΆΘθΘπΘπΘιΘδΘι Β»ΖΔ’ΙΝΥ‘ΎΕ© ικΡΒΡΧΦΕΥ–ό ΈΨΪΑ±ΥαΒΡΖΫΖ®ΘέΘ≥Θ±ΘίΓΘ Β―ιΫαΙϊ±μΟςΘ§ΧΦΕΥΨΪΑ±Υα–ό Έ≤ΜΫω‘ωΦ”ΝΥΕ© ικΡΒΡ ΠΝ ¬ί–ΐ≥ΧΕ»Θ§Εχ«“‘ω«ΩΝΥΕ© ικΡΒΡœΗΑϊ»κΡΛ–‘Θ§≤Δ«“ΈόΟςœ‘ΒΡœΗΑϊΕΨ–‘ΓΘ
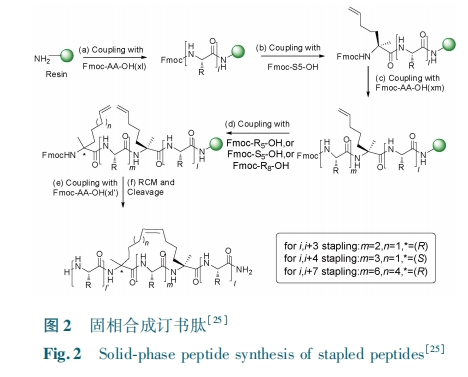
Θ≤.Θ¥ ≥ΛΝ¥Ε© ικΡΒΡΚœ≥…
“ΜΑψ»œΈΣΘ§”… Θ≥ΘΑ Ηω“‘…œΑ±ΜυΥα≤–ΜυΉι≥…ΒΡΕ© ικΡΒΡΚœ≥…≤ΜΡήΆ®ΙΐΙΧœύΚœ≥…“Μ¥ΈΗΏ–ßΚœ≥…Θ§Εχ–η“ΣΕύκΡΤ§ΕΈΝ§Ϋ”Ζ¥”Π Βœ÷ΓΘΘ≤ΘΑΘ±Θ≥ ΡξΘ§ Θ–ΘεΘνΘτΘεΘλΘθΘτΘε ΩΈΧβΉιΘ§ ”Ο ΓΑ Ή‘ »Μ Μ· ―ß Ν§ Ϋ” Ζ¥ ”Π Γ± Θ® ΘνΘαΘτΘιΘωΘε ΘψΘηΘεΘμΘιΘψΘαΘλΘλΘιΘγΘαΘτΘιΘοΘνΘ§ΘΈΘΟΘΧΘ© ΒΡΖΫΖ®Κœ≥…ΝΥ≥ΛΝ¥Ζ«Ψ≠ΒδΒΡΕ© ικΡΘ®Τδ≤ύΝ¥ΈΣΑκκΉΑ±Υα”κΝυΖζΜ·±ΫΜρ °ΖζΝΣ±ΫΖ¥”ΠΙΊΜΖΘ§ΕχΖ« Θ÷ΘεΘρΘδΘιΘνΘε –ΆΕ© ικΡΘ©¥”Εχ ΙΕ© ικΡΒΡΒΟΒΫΗϋΙψΖΚΒΡ”Π”ΟΘέΘ±ΘΙΘίΓΘΥϊΟ«≤…”ΟΒΡΝ§Ϋ”ΖΫΖ®ΈΣ ΘΥΘεΘνΘτ ΖΔΟςΒΡ ΘΈΘΟΘΧΘέΘ±ΘίΘ§Υϋ…φΦΑΒΣΕΥΑκκΉΑ±ΥακΡΕΈ”κΧΦΕΥΝρθΞκΡΕΈΓΘΘΈΘΟΘΧ ‘ΎΒΑΑΉ÷ Κœ≥…”κΑκΚœ≥…÷–»ΓΒΟΝΥΙψΖΚΒΡ”Π”ΟΘ§ΒΪ «ΧΦΕΥΝρθΞΒΡ ΘΤΘμΘοΘψ Μ·―ßΚœ≥…ΜΙ¥φ‘ΎΧτ’ΫΓΘΫϋΤΎΒΡ“ΜΗωΗΡΫχΖΫΖ® «Έ“Ο«ΖΔ’ΙΒΡάϊ”ΟΧΦΕΥθΘꬥζΧφΝρθΞΫχ––ΕύκΡθΘκ¬Ν§Ϋ”ΦΦ θΘέΘΕΘίΘ§ΗΟΖΫΖ®Μυ±Ψ‘≠άμ «‘Ύ»θΥαΧθΦΰœ¬ΕύκΡθΘꬱ̯«œθΥα―θΜ·ΈΣθΘΜυΒΰΒΣΘ§≤Δ‘ΎΖΦΜυΝρ¥Φ¥φ‘Ύœ¬‘≠ΈΜΉΣΜ·ΈΣΝρθΞΚσ”κ ΘΈ ΕΥΘΟΘυΘσ κΡ Βœ÷Μ·―ß―Γ‘ώ–‘Ν§Ϋ”Ζ¥”ΠΓΘ”κΝρθΞœύ±»Θ§ΧΦΕΥθΘκ¬κΡΩ…“‘÷±Ϋ”≤…”Ο ΘΤΘμΘοΘψ Ζ®ΙΧœύΚœ≥…ΗΏ–ßΒΟΒΫΘ§«““Ή”ΎΉ‘Ε·Μ·ΓΘ¥ΥΆβΘ§θΘκ¬κΡΩ…“‘ Βœ÷…ζΈο±μ¥οΘ§ ΙθΘκ¬Ζ®”Π”ΟΖΕΈßΗϋΦ”ΙψΖΚΓΘ
Θ≥ Ε© ικΡ…ζΈοΈοάμ–‘÷ ΒΡ±μ’ς
Ε© ικΡΒΡΫαΙΙΚΆ…ζΈοΜν–‘Ω…“‘Ά®ΙΐΕύ÷÷…ζΈοΈοάμΒΡ Β―ιΫχ––±μ’ςΘ§άΐ»γΘ§‘≤Εΰ…ΪΙβΤΉΘέΘ≥Θ≤ΘίΓΔΕΰΈ§ΚΥ¥≈Ι≤’ώΤΉΓΔΘΊ …δœΏΒΞΨß―ή…δΓΔ”ΪΙβΤΪ’ώΚΆ±μΟφΒ»άκΉ”ΧεΙ≤’ώΒ»ΓΘΈ“Ο«¥”“‘œ¬ ΘΒ ΗωΖΫΟφά¥≤ϊ ωΗΟΖΫΟφΒΡΫχ’ΙΓΘ
Θ≥.Θ± Ε© ικΡΫαΙΙΒΡ±μ’ς
‘≤Εΰ…ΪΙβΤΉΓΔΚΥ¥≈Ι≤’ώΤΉΚΆ ΘΊ …δœΏΒΞΨß―ή…δΨυΩ…“‘”Οά¥±μ’ςΕ© ικΡΒΡΕΰΦΕΫαΙΙΓΘ‘≤Εΰ…ΪΙβΤΉΩ…”Οά¥±μ’ςΕ© ικΡΒΡ ΠΝ ¬ί–ΐΒΡΈ»Ε®≥ΧΕ»ΓΘ‘ΎΕύκΡ÷–÷ς“ΣΒΡΙβΜν–‘ΜυΆ≈ «κΡΝ¥Ι«Φή÷–ΒΡκΡΦϋΓΔΑ±ΜυΥαΒΡΖΦœψ≤–ΜυΚΆΕΰΝρΦϋΓΘ»γΙϊΕύκΡΚ§”–≤ΜΙφ‘ρΒΡΕΰΦΕΫαΙΙΘ§Ρ«Ο¥Τδ‘≤Εΰ…ΪΙβΤΉ‘Ύ Θ±ΘΙΘΒ ΘνΘμ¥ΠΫΪ”–“ΜΗω«ΩΒΡΈϋ ’ΖεΘΜ»γΙϊΕύκΡΚ§”– ΠΝ ¬ί–ΐΫαΙΙΘ§Ρ«Ο¥Τδ‘≤Εΰ…ΪΙβΤΉ‘Ύ Θ≤ΘΑΘΗ ΘνΘμ ΚΆ Θ≤Θ≤Θ≤ ΘνΘμ ¥ΠΨυ”–Έϋ ’ΖεΓΘ“―÷ΣΕύκΡΒΡ≈®Ε»Θ§Ω…“‘ΗυΨί‘≤Εΰ…ΪΙβΤΉ‘ΎΘ≤Θ≤Θ≤ ΘνΘμ ΒΡΈϋ ’–≈Κ≈ά¥Ε®ΝΩΕ© ικΡΒΡ ΠΝ ¬ί–ΐ≥ΧΕ»ΓΘΆ®ΙΐΕ‘Ε© ικΡ‘Ύ≤ΜΆ§Έ¬Ε»œ¬ΒΡ‘≤Εΰ…ΪΙβΤΉΒΡ≤β ‘Θ§Ω…“‘÷±Ιέ―–ΨΩΕύκΡΙΙœσΒΡ»»Έ»Ε®–‘ΓΘΘ¬ΘαΘεΘκ Β»”Ο‘≤Εΰ…ΪΤΉ±μ’ςΝΥ Θ”ΘΝΘ»⁃ΘπΘΒΘ≥⁃ΘΗ ΒΡΫαΙΙΘ§ΖΔœ÷ΝΥ÷ßΦήΫαΙΙΡήΈ»Ε®ΕύκΡΒΡ ΠΝ ¬ί–ΐΘέΘ≥Θ≥ΘίΓΘ
ΚΥ¥≈Ι≤’ώΤΉΚΆ ΘΊ …δœΏΒΞΨß―ή…δΩ…“‘÷±Ϋ”ΡΘΡβ≥ωΕ© ικΡΒΡ ΒΦ ΫαΙΙΘ§≤ΔΩ…“‘»ΖΕ®Ε© ικΡ”κΑ–ΒΑΑΉΒΡΫαΚœΈΜΒψΘ§ΈΣΫχ“Μ≤ΫΒΡΕ© ικΡΒΡ…ηΦΤΧαΙ©ΝΥΫαΙΙΜυ¥ΓΓΘΘ–ΘηΘιΘλΘλΘιΘπΘσ Β»…ηΦΤΝΥΑ–œρ¥ΤΦΛΥΊ ήΧεΒΡΕ© ικΡΘέΘ≥Θ¥ΘίΓΘΆ®ΙΐΕ‘“ΜœΒΝ–ΥχΕ®ΕύκΡΒΡΚΥ¥≈”κΒΞΨß―ή…δΫαΙΙΒΡ±μ’ςΘ§ΥϊΟ«ΖΔœ÷Ε© ικΡΒΡΧΦΧΦ÷ßΦήΜα”ΑœλΤδ”κ ηΥ°ΒΑΑΉ±μΟφΒΡœύΜΞΉς”ΟΓΘ
Θ≥.Θ≤ Ε© ικΡ”κΑ–ΒΑΑΉΫαΚœΡήΝΠ±μ’ς
”ΪΙβΤΪ’ώ”κ±μΟφΒ»άκΉ”Ι≤’ώΦΦ θΩ…“‘”Ο”ΎΕύκΡ”κΑ–œρΒΑΑΉΫαΚœΡήΝΠΒΡ≤βΕ®Θ§≤Δ«“ΡήΕ®ΝΩ≤βΝΩΕ© ικΡΫαΚœ≥Θ ΐΘέΘ≥ΘΒ ΓΪ Θ≥ΘΗΘίΓΘ
ΤΪ’ώ”ΪΙβΒΡ«Ω»θ≥ΧΕ»”κ”ΪΙβΖ÷Ή”ΒΡ¥σ–Γ≥ ’ΐœύΙΊΘ§”κΤδ ήΦΛΖΔ ±ΉΣΕ·ΥΌΕ»≥ Ζ¥œύΙΊΓΘ‘ΎΙΧœύΕύκΡΚœ≥…ΒΡΙΐ≥Χ÷–ΫΪ“ΜΗω”ΪΙβΜυΆ≈“ΐ»κΕ© ικΡΘ§»γ“λΝρ«ηΥα”ΪΙβΥΊΓΘ‘Ύ»ή“Κ÷–Θ§Α–œρΒΑΑΉ”κΕ© ικΡΫαΚœΘ§Ω…“‘≤ζ…ζ”ΪΙβΤΪ’ώΘ§”ΪΙβΤΪ’ώ≥ΧΕ»”κΕύκΡΑσΕ®‘ΎΑ–œρΒΑΑΉΒΡ ΐΝΩ”–ΙΊΓΘΆ®Ιΐ≤βΝΩΤΪ’ώ”ΪΙβΘ§Ω…“‘Ε®ΝΩ≤βΕ®ΕύκΡ”κΑ–œρΒΑΑΉΒΡΫαΚœ≥Θ ΐΓΘΝμ“Μ÷÷ΗΡΫχΒΡ”ΪΙβΤΪ’ώΖΫΖ®Ω…“‘≤βΕ®≤ΜΚ§”ΪΙβΜυΆ≈ΒΡΕ© ικΡΉηΕœΒΑΑΉ⁃ΒΑΑΉœύΜΞΉς”ΟΒΡΡήΝΠΓΘ‘Ύ’β÷÷ΗΡΫχΒΡ”ΪΙβΤΪ’ώ÷–Θ§Κ§”–”ΪΙβ–ό ΈΒΡœΏ–‘Χλ»ΜΕύκΡ”κ≤ΜΚ§”ΪΙβ–ό ΈΒΡΕ© ικΡΜλΚœ‘Ύ“ΜΤπΓΘΆ®ΙΐΕ‘Κ§”–”ΪΙβ±ξΦ«ΒΡΧλ»ΜΕύκΡ”ΪΙβΤΪ’ώΫΒΒΆ≥ΧΕ»ΒΡ≤βΝΩΘ§ά¥≤βΝΩΕ© ικΡΉηΕœΧλ»ΜΕύκΡ”κΒΑΑΉΑ–œρΒΡΫαΚœΡήΝΠΓΘΘ≤ΘΑΘΑΘΙ ΡξΘ§Θ¬ΘαΘθΘτΘιΘσΘτΘαΒ»ΘέΘ≥ΘΙΘί”Ο”ΪΙβΤΪ’ώΒΡΖΫΖ®Θ§Ζ÷ΈωΝΥΙΙœσΥχΕ®ΒΡ Π¬ ΕύκΡ”κ ΘηΘΡΘΆΘ≤ ÷°ΦδΒΡΫαΚœΡήΝΠΓΘΥϊΟ«ΖΔœ÷ΙΙœσΥχΕ®ΒΡ Π¬ΕύκΡ”κΑ–ΒΑΑΉΒΡΫαΚœΡήΝΠ”κ“ΐ»κΙΙœσΥχΕ®÷ßΦήΒΡΈΜ÷ΟœύΙΊΘ§”––©ΈΜ÷ΟΩ…“‘Οςœ‘‘ω«ΩΫαΚœΡήΝΠΘ§”––©ΈΜ÷Ο»¥ΜαΟςœ‘ΫΒΒΆΫαΚœΡήΝΠΓΘ
‘Ύ±μΟφΒ»άκΉ”ΧεΙ≤’ώ Β―ι÷–Θ§ Ήœ»ΫΪ…ζΈοΥΊ–ό ΈΒΡΕ© ικΡΙΧΕ®‘Ύ…ζΈο¥ΪΗ––ΨΤ§…œΘ§»ΜΚσΫΪΤ䱩¬Ε”ΎΑ–œρΒΑΑΉ÷–ΓΘ’β―υΕ© ικΡ”κΑ–œρΒΑΑΉ÷°ΦδΒΡœύΜΞΉς”ΟΩ…“‘±Μ÷±Ϋ”≤βΕ®Θ§≤Δ«“Ω…“‘ΒΟΒΫΫαΚœΒΡΕ·ΝΠ―ßΙΐ≥ΧΓΘ±μΟφΒ»άκΉ”Χε Β―ι“≤Ω…“‘”Ο”Ύ≤βΕ®Ε© ικΡ“÷÷ΤΒΑΑΉΗ¥ΚœΈο–Έ≥…ΒΡ≥ΧΕ»ΓΘΫΪΒΑΑΉΗ¥ΚœΈοΙΧΕ®‘Ύ…ζΈο¥ΪΗ––ΨΤ§…œΘ§»ΜΚσΫΪΤ䱩¬Ε‘ΎΕ© ικΡ÷–Θ§’β―υΩ…“‘≤βΕ®Ε© ικΡΕ‘ΕύΒΑΑΉΗ¥ΚœΈοΒΡΫαΚœ”κΫβάκΕ·ΝΠ―ß”ΑœλΓΘ
Θ≥. Θ≥ Ε© ικΡΟΗΫβΈ»Ε®–‘ΒΡ±μ’ς
¥”»ΥΙΛΚœ≥…ΕύκΡΒΫ“©ΈοΘ§ΕύκΡΒΡΟΗΫβΈ»Ε®–‘≥…ΈΣ“ΜΗω÷Ί“ΣΒΡ÷Η±ξΘ§“≤ «“ΜΗω÷ς“ΣΒΡ’œΑ≠ΓΘ”…”ΎΒΑΑΉΥ°ΫβΟΗΥ°ΫβθΘΑΖΦϋ–η“ΣΕύκΡ”–“ΜΗω φ’ΙΒΡΙΙœσΘ§“ρ¥Υ”Ο÷ßΦήά¥ΥχΕ®ΕύκΡΒΡ ΠΝ ¬ί–ΐΫαΙΙΩ…“‘”––ßΒΊ“÷÷ΤΕύκΡ±ΜΒΑΑΉΥ°ΫβΟΗΥ°ΫβΓΘΈΣΝΥΤάΙάΕύκΡΒΡΧεΆβΟΗΫβΈ»Ε®–‘Θ§–η“ΣΜυ”ΎΕ© ικΡΒΡ–ρΝ–―Γ‘ώΒΑΑΉΥ°ΫβΟΗΓΘ“»ΒΑΑΉΟΗΩ…“‘ Ε±πΨΪΑ±Υα Θ® ΘΝΘρΘγΘ© ΚΆ άΒ Α± ΥαΘ®ΘΧΘυΘσΘ©Θ§Ο”ΒΑΑΉΟΗ÷ς“Σ Ε±π±Ϋ±ϊΑ±ΥαΘ®Θ–ΘηΘεΘ©ΓΔά“Α±Υα(Θ‘ΘυΘρΘ©ΓΔΝΝΑ±ΥαΘ®ΘΧΘεΘθΘ©ΚΆΦΉΝρΑ±ΥαΘ®ΘΆΘεΘτΘ©ΓΘΘ¬ΘιΘρΘδ Β»ΘέΘ¥ΘΑΘί±μ’ςΝΥ Θ¬ΘψΘλ⁃Θ≤ ”ρΒΡΕ© ικΡΘ§≤ΔΫχ––ΝΥΟΗΫβΈ»Ε®–‘ΒΡ≤βΕ®ΓΘΥϊΟ«Ά®ΙΐΕ‘±»”ΪΙβΥΊ±ξΦ«ΒΡΕ© ικΡ”κ”ΪΙβΥΊ±ξΦ«ΒΡœΏ–‘κΡ‘Ύ“»ΒΑΑΉΟΗ¥φ‘Ύœ¬≤ΜΆ§ ±ΦδΕΈΒΡ”ΪΙβ«ΩΕ»±δΜ·ά¥»ΖΕ®Ε© ικΡΒΡΟΗΫβΈ»Ε®–‘ΓΘΕ‘Ε© ικΡΫχ––ΧεΡΎΜρΧεΆβΒΡ―Σ«εΈ»Ε®–‘≤βΕ®Ω…“‘ΗϋΦ”Ος»ΖΒΊ±μΟςΕύκΡΒΡΟΗΫβΈ»Ε®–‘ΓΘ
Θ≥.Θ¥ Ε© ικΡ»κΡΛ–‘ΒΡ±μ’ς
Νς ΫœΗΑϊΦΤΚΆΙ≤ΨέΫΙ”ΪΙβœ‘ΈΔΨΒ’βΝΫ÷÷ΖΫΖ®Ω…“‘άϊ”Ο”ΪΙβ±ξΦ«Ε© ικΡά¥―–ΨΩΕύκΡΒΡœΗΑϊΆ®ΆΗ–‘ΘέΘ¥Θ±ΘίΓΘΒΞΗωΚ§”–”ΪΙβΖ÷Ή”ΒΡœΗΑϊΩ…“‘±ψΫίΒΊ±ΜΝς ΫœΗΑϊΦΤ…Η―Γ≥ωΘ§’β―υΩ…“‘ΨΪ»ΖΝΥΫβΕ© ικΡΒΡ»κΡΛ–ßΙϊΓΘΒΪ «Θ§Νς ΫœΗΑϊΦΤ ΐ≤ΜΡήΙέ≤λΕ© ικΡ‘ΎœΗΑϊ÷–ΒΡΨΏΧεΈΜ÷ΟΓΘ”…”ΎπΛΗΫ‘ΎœΗΑϊ±μΟφΒΡΕ© ικΡΜα≤ζ…ζ»κΡΛΒΡ¥μΈσ–≈œΔΘ§‘ΎΖ÷ΈωΕ© ικΡ»κΡΛ–‘ ±Θ§±Ί–κ”Ο“»ΒΑΑΉΟΗ≥ΐ»ΞΈϋΗΫœΗΑϊ±μΟφΒΡΕ© ικΡΓΘΙ≤ΨέΫΙ”ΪΙβœ‘ΈΔΨΒΖ®―–ΨΩΒΡœΗΑϊ ΐΝΩΟςœ‘“Σ…Ό”ΎΝς ΫœΗΑϊΦΤΥυ―–ΨΩΒΡ ΐΡΩΓΘ–η“Σ÷Η≥ωΒΡ «Θ§Ι≤ΨέΫΙ”ΪΙβœ‘ΈΔΨΒΖ®Ω…“‘―–ΨΩΕ© ικΡΒΡ»κΡΛΒΡΗϋΕύœΗΫΎΘ§»γΕ© ικΡ‘ΎœΗΑϊΡΎΒΡΨΏΧεΈΜ÷ΟΘ§ΒΪ «Ι≤ΨέΫΙœ‘ΈΔΨΒΖ®÷ΜΡήΕ®–‘ΒΡ≈–ΕœΕύκΡ «ΖώΡήΫχ»κœΗΑϊΓΘΉνΫϋΖΔ’ΙΝΥ“Μ÷÷Ω…“‘Ε®ΝΩ≤βΕ®ΕύκΡ»κΡΛ–‘ΒΡ”ΪΙβœ‘ΈΔΨΒΖ®Θ§“≤Ω…“‘÷±Ϋ”Ε®ΝΩ±»ΫœΕύκΡΒΡ»κΡΛ–‘≤ν“λΘ§ΒΪ≤ΜΡήΧαΙ©Ω… ”Μ·–≈œΔΓΘ
ΘΆΘθΘπΘπΘιΘδΘι Β»ΘέΘ≥Θ±Θί≤…”ΟΝς ΫœΗΑϊΦΤΚΆΙ≤ΨέΫΙ”ΪΙβœ‘ΈΔΨΒ―–ΨΩΝΥΨΪΑ±Υα–ό ΈΒΡΕ© ικΡΒΡ»κΡΛΙΐ≥ΧΓΘΆ®ΙΐΝς ΫœΗΑϊΦΤΒΡΖΫΖ®Θ§ΥϊΟ«ΖΔœ÷ΨΪΑ±Υα–ό ΈΒΡΕ© ικΡΩ…“‘Οςœ‘ΧαΗΏΕύκΡΒΡ»κΡΛ–‘ΘΜάϊ”ΟΙ≤ΨέΫΙœ‘ΈΔΨΒΖ®Θ§ΥϊΟ«Ϋχ“Μ≤ΫΒΡ»Ζ»œΝΥΗΟΫαΙϊΘ§≤ΔΧα≥ωΨΪΑ±Υα–ό ΈΒΡΕ© ικΡΒΡ»κΡΛΩ…Ρή «Ά®ΙΐΡΎΆΧΜζάμ Βœ÷ΒΡΓΘ
Θ≥.ΘΒ Ε© ικΡ…ζΈοΜν–‘ΒΡ±μ’ς
άμ¬έΦΤΥψΫαΙΙ±μΟςΕ© ικΡ÷–÷ßΦήΒΡΨ÷≤ΩΈ»Ε®Ής”Ο±» ΠΝ ¬ί–ΐ≥ΧΕ»Ε‘…ζΈοΜν–‘ΒΡ”ΑœλΩ…ΡήΗϋ¥σΘ§≤βΕ®Ε© ικΡΒΡ…ζΈοΜν–‘“≤≥…ΈΣ±Ί≤ΜΩ……ΌΒΡ“Μ≤ΫΓΘΧεΆβΟβ“ΏΙ≤≥ΝΒμΩ…“‘≤βΕ®Ε© ικΡ‘ΎœΗΑϊΜΖΨ≥œ¬”κΑ–ΒΑΑΉΒΡΫαΚœΡήΝΠΘέΘ¥Θ≤ ΓΪ Θ¥ΘΒΘίΓΘΕ© ικΡΒΡΟβ“ΏΙ≤≥ΝΒμΙΐ≥ΧΩ…“‘Φρ ωΈΣ”ΟΩΙΧεΫΪΡΩ±ξΒΑΑΉΧΊ“λ–‘≥ΝΒμœ¬ά¥Θ§Ά§ ±”κΡΩ±ξΒΑΑΉΫαΚœΒΡΕ© ικΡ“≤“ΜΤπ±Μ≥ΝΒμΓΘ≥Θ”ΟΝΫ÷÷Ε© ικΡΫχ––ΗΟ Β―ιΘ§“Μ÷÷ «άϊ”Ο”ΪΙβΥΊ±ξΦ«ΒΡΕ© ικΡΘ§Νμ“Μ÷÷ «άϊ”Ο…ζΈοΥΊ±ξΦ«ΒΡΕ© ικΡΓΘΨΏΧε≤Ϋ÷η÷ς“ΣΈΣΘ§‘ΎœΗΑϊΝ―Ϋβ“Κ÷–Φ”»κΑ–ΒΑΑΉΒΡΧΊ“λ–‘ΩΙΧεΘ§Ζθ”ΐ“ΜΕΈ ±ΦδΚσΘ§Φ”»κ”κΩΙΧεΧΊ“λ–‘ΫαΚœΒΡ«μ÷§Χ«…œΒΡΒΑΑΉ ΘΝ ΜρΒΑΑΉ Θ«ΓΘΒ±Ε© ικΡ”κΑ–ΒΑΑΉΫαΚœ ±Θ§Ε© ικΡ“≤Ά§ ±±Μ≥ΝΫΒΘΜ≥ΝΒμΗ¥ΚœΈοΆ®Ιΐ±δ–‘Ψέ±ϊœ©θΘΑΖΡΐΫΚΒγ”Ψ”÷±ΜΖ÷ΩΣΘΜΉνΚσ≤…”ΟΟβ“Ώ”ΓΦΘ Β―ιΫχ“Μ≤Ϋ÷Λ ΒΙΙœσΥχΕ®κΡ”κΑ–ΒΑΑΉΫαΚœΓΘ
Ε© ικΡ «ΖώΨΏ”–«±‘ΎΒΡ“Ϋ―ß”Π”Ο «Ά®ΙΐΜνΧεΡΎΤδΙΠΡήΒΡ≤βΝΩά¥ΤάΙάΒΡΓΘΫΪΕ© ικΡΉΔ»κΒΫΜνΒΡΜΦ≤ΓΕ·ΈοΧεΡΎΘ§Ιέ≤λΚΆ≤βΕ®Ε·ΈοΧεΡΎΗςœν…ζάμ÷Η±ξ“‘»ΖΕ®Ε© ικΡΒΡ…ζΈοΜν–‘ΓΘ
Θ¥ Ε© ικΡΒΡ…ζΈο“Ϋ―ßΙΠΡή”κ”Π”Ο
ΒΫΡΩ«ΑΈΣ÷ΙΘ§ΙζΦ …œ”––μΕύΩΈΧβΉιΕΦ‘Ύ¥” ¬”κΕ© ικΡœύΙΊΒΡ―–ΨΩΘ§≤Δ»ΓΒΟΝΥ“ΜΕ®ΒΡ≥…ΙϊΓΘΕ© ικΡ‘Ύ÷ΈΝΤΑ©÷ΔΓΔ“÷÷ΤΑ§ΉΧ≤ΓΚΆ±ϊ–ΆΗΈ―Ή“‘ΦΑΒςΫΎ–≈Κ≈Ά®¬ΖΒ»ΖΫΟφΒΡ”Π”ΟΨυ”–±®ΒάΓΘœ¬Οφ÷ς“ΣΫΪ¥”’βΦΗΗωΖΫΟφΦρΒΞΫι…ή“Μœ¬Ε© ικΡΒΡ“Ϋ―ß”Π”ΟΓΘ
Θ¥. Θ± Ε© ικΡ‘ΎΑ©≤ΓΖά÷Έ÷–ΒΡ”Π”Ο
Θ¥.Θ±.Θ± ΒςΩΊ ΘπΘΒΘ≥ Μυ“ρ
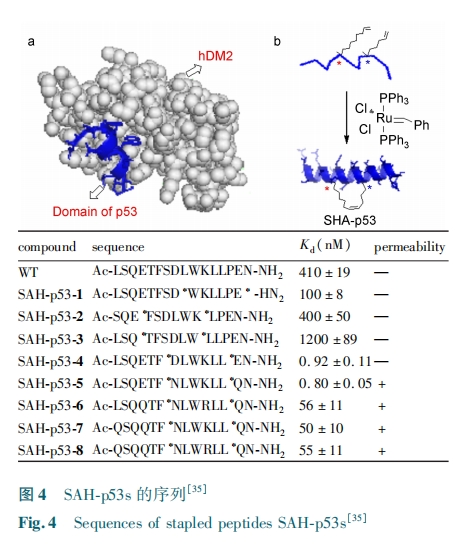
ΘπΘΒΘ≥ Μυ“ρ «“Μ÷÷“÷Α©Μυ“ρΘ§ «œΗΑϊ…ζ≥Λ÷ήΤΎ÷–ΒΡΗΚΒςΫΎ“ρΉ”Θ§”κ ΘΡΘΈΘΝ ΒΡ–όΗ¥ΓΔœΗΑϊΖ÷Μ·ΓΔœΗΑϊ÷ήΤΎΒΡΒςΩΊΒ»…ζΟϋΙΐ≥Χ”–ΙΊΓΘΘπΘΒΘ≥ ΒΡ»± ßΚΆΆΜ±δΦΑœύΙΊΒΑΑΉΟΗΧεΒΡΫΒΫβΜαΒΦ÷¬÷ΉΝωœΗΑϊΒΡ≤ζ…ζΓΘ–όΗ¥ ΘπΘΒΘ≥ΒΡΜν–‘Ω…“‘ΗϋΦ””––ßΒΊ÷ΈΝΤΑ©÷ΔΓΘΘ≈Θ≥ ΖΚΥΊΝ§Ϋ”ΟΗΘηΘΡΘΆΘ≤ ÷±Ϋ”ΫαΚœ‘Ύ ΘπΘΒΘ≥ ΒΑΑΉΒΡΖ¥ ΫΦΛΜν”ρΘ§Α–œρ ΘπΘΒΘ≥ΒΑΑΉΫχ––ΟΗΫβ»γΆΦ Θ¥Θα Υυ ΨΓΘΘηΘΡΘΆΘ¥ Θ· ΘηΘΡΘΆΘχ Ω…“‘“÷÷ΤΘπΘΒΘ≥ ΒΡΜν–‘ΓΘ÷ΈΝΤΑ©÷ΔΒΡ“ΜΗω”––ßΒΡΖΫΖ® «―Α’““Μ÷÷Ω…“‘“÷÷Τ ΘηΘΡΘΆΘ≤ Θ· ΘηΘΡΘΆΘχ ”κ ΘπΘΒΘ≥ ΫαΚœΒΡΜν–‘Ζ÷Ή”ΓΘΜυ”Ύ ΘπΘΒΘ≥ –ρΝ–Εχ…ηΦΤΒΡΕύκΡάύ“÷÷ΤΦΝΩ…“‘ΗΏ–ßΒΊΫαΚœ ΘηΘΡΘΆΘ≤ΘΜΒΪœΏ–‘ΒΡΕύκΡ≤ΜΡήΫχ»κœΗΑϊΡΛΘ§«“ΟΗΈ»Ε®–‘≤νΓΘΜυ”Ύ¥ΥΘ§ Θ¬ΘεΘρΘνΘαΘλ Β»ΘέΘ≥ΘΒΘίάϊ”ΟΧΦΧΦ÷ßΦή…ηΦΤΚœ≥…ΙΙœσΥχΕ®κΡΘ§Οςœ‘ΧαΗΏΝΥΕύκΡΒΡ»κΡΛ–‘ΚΆΟΗΫβΈ»Ε®–‘Θ§Ε© ικΡΒΡΫαΙΙ»γΆΦ Θ¥Θβ ”κ Θ¥Θψ Υυ ΨΓΘΚ§”– Θ±ΘΒ ΗωΑ±ΜυΥαΒΡΙΙœσΥχΕ®Ω…“‘“÷÷Τ ΘπΘΒΘ≥ ΒΑΑΉ”κ ΘηΘΡΘΆΘ≤ ΒΑΑΉ÷°ΦδΒΡœύΜΞΉς”ΟΓΘΕ© ικΡΒΡ ΠΝ ¬ί–ΐΫαΙΙΜα≤ε»κΒΫΘηΘΡΘΆΘ≤ ±μΟφΒΡ ηΥ°ΑΦ≤έ÷–¥”Εχ“÷÷Τ ΘπΘΒΘ≥ ”κ÷°ΫαΚœΓΘΗΟΕ© ικΡ÷–ΒΡ»ΐΗωΑ±ΜυΥαΕ‘”ΎΫαΚœ ΘηΘΡΘΆΘ≤ «±Ί≤ΜΩ……ΌΒΡΘ§Ζ÷±π « Θ±ΘΙ ΈΜ±Ϋ±ϊΑ±ΥαΓΔΘ≤Θ≥ ΈΜ…ΪΑ±ΥαΚΆ Θ≤ΘΕ ΈΜΝΝΑ±ΥαΓΘΘ”ΘΝΘ»⁃ΘπΘΒΘ≥ ΝΤΖ®Ά®ΙΐΙΙœσΥχΕ®κΡ Θ”ΘΝΘ» Α–œρΉΣ¬ΦΙΐ≥ΧΦΛΜν ΘπΘΒΘ≥ ¥οΒΫ“÷÷Τ÷ΉΝωΓΘΗΟ≤Ώ¬‘ΒΡ…ηΦΤΥΦ¬Ζ»γœ¬Θ§ Ήœ»Θ§…ηΦΤ”κ ΘηΘΡΘΆΘ≤ œύΜΞΉς”ΟΒΡΕ© ικΡΘ§ΧΦΧΦ÷ßΦήΒΡΈΜ÷ΟΨΓΝΩ±ήΩΣ”κ ΘηΘΡΘΆΘ≤ ΫαΚœΒΡ÷Ί“Σ≤ΩΈΜΘΜΤδ¥ΈΘ§άϊ”Οœ©ΧΰΗ¥Ζ÷ΫβΖ¥”ΠΚœ≥…ΝΥ Θ¥ Χθ ΘιΘ§Θι ΘΪ ΘΖ ΈΜΕ© ικΡΘ®Θ”ΘΝΘ»⁃ΘπΘΒΘ≥Θσ Θ± ΓΪ Θ¥Θ©ΘΜΉνΚσΘ§Ε‘Ε© ικΡ Θ”ΘΝΘ»⁃ΘπΘΒΘ≥Θσ Θ±ΓΪ Θ¥ Ϋχ––…ζΈοΈοάμ Β―ι±μ’ςΓΘ‘≤Εΰ…ΪΤΉ±μΟςΕύκΡΒΡΠΝ ¬ί–ΐ≥ΧΕ»ΕΦ”–Υυ‘ωΦ”ΘΜ”ΪΙβΤΪ’ώ Β―ιΖΔœ÷Ε© ικΡΒΡ«ΉΚΆΝΠ”κΧλ»ΜΕύκΡœύΫϋΓΘœΗΑϊ Β―ιΖΔœ÷ Θ”ΘΝΘ»⁃ΘπΘΒΘ≥Θσ Θ± ΓΪ Θ¥ ΨυΡ―“‘ΩγΙΐœΗΑϊΡΛΓΘ‘Ύ…ζάμ ΘπΘ» ΧθΦΰœ¬Θ§Ε© ικΡ Θ”ΘΝΘ»⁃ΘπΘΒΘ≥Θσ Θ± ΓΪ Θ¥ ”…”Ύ¥χΝΫΗωΗΚΒγΚ…ΕχΡ―“‘Ά®ΙΐœΗΑϊΡΛΓΘΥϊΟ«ΫΪΧλΕ§Α±Υα”κΙ»Α±ΥαΖ÷±πΧφΜΜΈΣΧλΕ§θΘΑΖ”κΙ»Α±θΘΑΖΘ§ΖΔ’ΙΝΥΒΎΕΰ¥ζΕύκΡΜ·ΚœΈο Θ”ΘΝΘ»⁃ΘπΘΒΘ≥Θσ ΘΒ ΓΪ ΘΗΓΘœΗΑϊ Β―ι±μΟς Θ”ΘΝΘ»⁃ΘπΘΒΘ≥ ΘΗ ”κ ΘηΘΡΘΆΘ≤ ”–Ή≈ΗΏΒΡ«ΉΚΆ–‘≤ΔΩ…“‘ΩγΙΐœΗΑϊΡΛΓΘΘΉΘεΘσΘτΘεΘρΘν ”ΓΦΘ Β―ιΫχ“Μ≤Ϋ÷Λ Β Θ”ΘΝΘΆ⁃ΘπΘΒΘ≥ ΘΗ ΡήΜ÷Η¥ ΘπΘΒΘ≥ ΒΡ’ΐ≥ΘΥ°ΤΫΓΘΗΟ―–ΨΩ±μΟςΕ© ικΡΩ…“‘”Ο”ΎΉηΕœΒΑΑΉ⁃ΒΑΑΉΓΘ
Θ≤ΘΑΘ±ΘΑ ΡξΘ§ Θ¬ΘεΘρΘνΘαΘλ Β»ΘέΘ¥ΘΕΘί Ε‘ Θ”ΘΝΘ»⁃ΘπΘΒΘ≥ ΘΗ ΒςΩΊ ΘπΘΒΘ≥Ϋχ–– ΝΥ Ϋχ “Μ ≤Ϋ ΒΡ ―– ΨΩΓΘΘηΘΡΘΆΘχ ΒΡ Ϋα ΙΙ Ζ÷ Έω ΖΔ œ÷ΘηΘΡΘΆΘχ “÷÷Τ ΘπΘΒΘ≥ ΒΡΖ¥ ΫΦΛΜνΒΡ ΠΝ ¬ί–ΐ”κ ΘηΘΡΘΆΘ≤ ΒΡΡΘ ΫΦΪΈΣœύΥΤΓΘ“ρΕχΘ§ΥϊΟ«≤β ‘ Θ”ΘΝΘ»⁃ΘπΘΒΘ≥ ΘΗ Ε‘ ΘηΘΡΘΆΘχΒΡΑ–œρ“÷÷ΤΉς”ΟΘ§≤Δ«“―–ΨΩΝΥ ΘηΘΡΘΆΘχ ±Μ“÷÷ΤΚσΒΡΧεΡΎ ΘπΘΒΘ≥ ΙΠΡή±δΜ·Θ§ Β―ιΫαΙϊ±μΟςΘ§Θ”ΘΝΘ»⁃ΘπΘΒΘ≥ ΘΗ Ω…“‘”––ßΒΊ“÷÷Τ ΘηΘΡΘΆΘχΓΘΘ”ΘΝΘ»⁃ΘπΘΒΘ≥ ΘΗ Φ»Ω…“‘Α–œρ ΘηΘΡΘΆΘ≤ “≤Ω…“‘Α–œρ ΘηΘΡΘΆΘχΘ§≤Δ«“ Θ”ΘΝΘ»⁃ΘπΘΒΘ≥ ΘΗ Ε‘”Ύ ΘηΘΡΘΆΘχ Α–œρΡήΝΠ±»Ε‘ ΘηΘΡΘΆΘ≤ ΒΡΑ–œρΡήΝΠΗΏ Θ≤ΘΒ ±ΕΓΘΘ”ΘΝΘ»⁃ΘπΘΒΘ≥ ΘΗ Α–œρœΗΑϊάοΒΡ ΘηΘΡΘΆΘχΘ§ΉηΕœ ΘπΘΒΘ≥⁃ΘηΘΡΘΆΘΊ Η¥ΚœΈοΒΡ–Έ≥…Θ§¥”ΕχΦΛΜν ΘπΘΒΘ≥ ΒΡ÷ΉΝω“÷÷ΤΆ®¬ΖΓΘ“ρ¥ΥΘ§Ε‘ ΘηΘΡΘΆΘ≤ ΚΆΘηΘΡΘΆΘχ ΥΪΑ–œρΒΡ“÷÷ΤΦΝΩ…“‘ΗϋΚΟΒΊ”Ο”Ύ÷ΈΝΤΑ©÷ΔΓΘ
Θ¥.Θ±.Θ≤ Ε‘”Ύ Θ¬ΘψΘλ⁃Θ≤ ΒΑΑΉΦ“ΉεΒΡΒςΩΊ
Θ¬ ΝήΑΆœΗΑϊΝω⁃Θ≤ Μυ“ρΘ®Θ¬⁃ΘψΘεΘλΘλ ΘλΘυΘμΘπΘηΘοΘμΘα⁃Θ≤Θ©Φρ≥ΤΘ¬ΘψΘλ⁃Θ≤Θ§ «ΒςΩΊœΗΑϊΒρΆωΒΡ÷Ί“ΣΜυ“ρ÷°“ΜΓΘΘ¬ΘψΘλ⁃Θ≤ Μυ“ρ «“Μ÷÷‘≠Α©Μυ“ρΘ§‘Ύ“÷÷ΤœΗΑϊΒρΆωΙΐ≥Χ÷–ΖΔΜ”÷Ί“ΣΒΡΉς”ΟΓΘΘ¬ΘψΘλ⁃Θ≤ Φ“ΉεΒΑΑΉ «”… Θ¥ Ηω±Θ ΊΒΡ Θ¬ΘψΘλ⁃Θ≤ Ά§‘¥ΒΡ«χ”ρΉι≥…Θ§«“ΟΩΗωΆ§‘¥”ρΕΦΑϋΚ§“ΜΗω ΠΝ ¬ί–ΐΫαΙΙΓΘΗυΨί‘ΎœΗΑϊΒρΆωΙΐ≥Χ÷–ΥυΤπΒΡΙΠΡή≤ΜΆ§Θ§Θ¬ΘψΘλ⁃Θ≤ΒΑΑΉΦ“ΉεΩ…“‘Ζ÷ΈΣΝΫ¥σάύΘ§“ΜάύΨΏ”–“÷÷ΤœΗΑϊΒςΆωΉς”ΟΘ§»γ Θ¬ΘψΘλ⁃Θ≤ΓΔΘ¬ΘψΘλ⁃ΘΊΘΧΓΔΘ¬ΘψΘλ⁃Θ±ΓΔΘΆΘψΘλ⁃Θ± Β»ΘΜΝμ“ΜάύΨΏ”–¥ΌΫχœΗΑϊΒρΆωΉς”ΟΘ§ »γ Θ¬ΘαΘχΓΔ Θ¬ΘψΘλ⁃ΘΊΘσΓΔ Θ¬ΘαΘκΓΔ Θ¬ΘιΘδ Β»ΓΘΘ¬ΘψΘλ⁃Θ≤ Φ“ΉεΒΑΑΉΙΙ≥…ΝΥ“ΜΗωΒςΫΎœΗΑϊΒρΆωΒΡΙΊΦϋΒψΓΘΉνΫϋ―–ΨΩΖΔœ÷ Θ¬ΘψΘλ⁃Θ≤ Φ“ΉεΒΑΑΉ≥ΐΝΥΒςΩΊœΏΝΘΧε÷–ΒρΆω“ρΉ”ΒΡ ΆΖ≈ΆβΘ§ΜΙ‘Ύ…ζΟϋΜνΕ·÷–Αγ―ί’Ώ–μΕύ–¬ΒΡΫ«…ΪΘ§άΐ»γΘ§ ΒςΫΎ–¬≥¬¥ζ–ΜΘέΘ¥ΘΖΘ§Θ¥ΘΗΘίΓΔΧεΡΎ ΘΟΘαΘ≤ ΘΪ ≈®Ε»ΘέΘ¥ΘΙΘίΚΆœΏΝΘΧε–ΈΧ§ΘέΘΒΘΑΘίΓΘΘ¬ΘψΘλ⁃Θ≤ ΒΑΑΉΆ®ΙΐΑϋΚ§Τδ÷–ΒΡ ΠΝ ¬ί–ΐΫαΙΙΒΡ Θ¬Θ»Θ≥ Τ§ΕΈά¥ΒςΫΎœΗΑϊΡΎΒΡΒΑΑΉ⁃ΒΑΑΉœύΜΞΉς”ΟΘ§Ήν÷’ΒςΫΎ–μΕύ…ζΈοΙΐ≥ΧΓΘ
Χλ»ΜœΏ–‘ΒΡΕΧκΡ≤ΜΡή±Θ≥÷‘≠”–ΕΰΦΕΫαΙΙΘ§Ρ―“‘Ϋχ»κœΗΑϊΡΛΘ§«“Ε‘ΒΑΑΉΥ°ΫβΟΗ≤ΜΈ»Ε®ΓΘΘ≤ΘΑΘΑΘ¥ ΡξΘ§ΘΉΘαΘλΘεΘνΘσΘκΘυ Β»ΘέΘ≥ΘΖΘί”ΟΧΦΧΦ÷ßΦήά¥Έ»Ε®ΒΡΕΧκΡΒΡΙΙœσΓΘΆ®ΙΐΡΘΡβ Θ¬ΘιΘδ ΒΡ Θ¬Θ»Θ≥ «χ”ρΘ§…ηΦΤΝΥ“ΜΉιΨΏ”–Έ»Ε® ΠΝ¬ί–ΐΫαΙΙΒΡΕ© ικΡ Θ”ΘΝΘ»Θ¬ΘσΘ§»γ±μ Θ≤ Υυ ΨΓΘΆ®ΙΐΕ‘Χλ»Μ Θ¬Θ»Θ≥ ”κ Θ”ΘΝΘ»Θ¬Θσ ΒΡ‘≤Εΰ…ΪΤΉ±»ΫœΘ§ΥϊΟ«ΖΔœ÷Χλ»ΜΕύκΡ÷ς“Σ“‘≤ΜΙφ‘ρΨμ«ζΒΡ–Έ Ϋ¥φ‘ΎΘ§÷Μ”– Θ±ΘΕΘΞ ΒΡ ΠΝ¬ί–ΐ≥ΧΕ»ΘΜΕχ Θ”ΘΝΘ»Θ¬Θσ ΒΡ ΠΝ ¬ί–ΐ≥ΧΕ»‘ρΧαΗΏΒΫ Θ≥ΘΒΘΞΒΫ ΘΗΘΖΘΞ ΓΘΟΗΫβΈ»Ε®–‘ΒΡ Β―ι÷Λ ΒΘ§”κΧλ»Μ Θ¬Θ»Θ≥ œύ±»Θ§Θ”ΘΝΘ»Θ¬Θσ ΨΏ”–ΗϋΗΏΒΡΟΗΫβΈ»Ε®–‘“‘ΦΑ―ΣΫ§Έ»Ε®–‘ΓΘ¥ΥΆβΘ§ΥϊΟ«ΜΙΆ®ΙΐΚΥ¥≈ Β―ιΖ÷±π―–ΨΩ Θ¬Θ»Θ≥ ΚΆΘ”ΘΝΘ»Θ¬ΘΝ”κ Θ¬ΘψΘλ⁃ΘΊΘΧ ΒΡœύΜΞΉς”ΟΓΘΆβΦ”Χλ»ΜΕύκΡ Θ¬ΘιΘδΘ¬Θ»Θ≥ Μρ’Ώ Θ”ΘΝΘ»Θ¬ΘΝ ”ΎΘ±ΘΒ ΘΈ ±ξΦ«ΒΡ Θ¬ΘΟΘΧ⁃ΘΊΘΧ »ή“ΚΚσΘ§≤…Φ· Θ¬ΘψΘλ⁃ΘΊΘΧ ΒΡΕΰΈ§Θ±ΘΒΘΈ⁃Θ±Θ» “λΚΥΒΞΝΩΉ”œύΙΊΚΥ¥≈Ι≤’ώΤΉΘ® Θ»Θ”Θ―ΘΟΘ©ΓΘΝΫ÷÷ Θ»Θ”Θ―ΘΟ ΤΉΆΦΦΪΈΣœύΥΤΘ§ ’βΥΒΟςΘ”ΘΝΘ»Θ¬ΘΝΚΆΧλ»ΜΕύκΡ Θ¬ΘιΘδ Θ¬Θ»Θ≥ ”κΑ–œρΒΑΑΉ Θ¬ΘψΘλ⁃ΘΊΘΧ ΒΡœύΜΞΉς”ΟΡΘ ΫœύΆ§ΓΘ”ΪΙβΤΪ’ώ Β―ιΖΔœ÷ Θ”ΘΝΘ»Θ¬ΘΝΫαΚœ Θ¬ΘψΘλ⁃ΘΊΘΧ ΒΡΡήΝΠ±»Χλ»ΜΕύκΡ Θ¬ΘιΘδ Θ¬Θ»Θ≥ ΗΏ ΘΕ ±ΕΓΘ―–ΨΩΖΔœ÷‘Ύ–Γ σΒΡΗΈ‘ύœΗΑϊ÷–Θ§Θ”ΘΝΘ»Θ¬ΘΝΜα ΙœΗΑϊ…ΪΥΊΘψ ΆΖ≈‘ωΦ”Θ§ΕχΧλ»ΜΕύκΡ Θ¬ΘιΘδ Θ¬Θ»Θ≥ Ε‘œΗΑϊ…ΪΥΊ Θψ ΆΖ≈ΝΩΦΗΚθΟΜ”–”ΑœλΓΘ’β“≤Ϋχ“Μ≤Ϋ÷Λ ΒΝΥ Θ”ΘΝΘ»Θ¬ΘΝΡήΙΜΦΛΜνœΗΑϊΒρΆω–≈Κ≈Ά®¬ΖΓΘΥφΚσΒΡœΗΑϊ»κΡΛ Β―ι÷ΛΟςΘ”ΘΝΘ»Θ¬ΘΝΩ…“‘Ϋχ»κœΗΑϊΡΛΓΘ…œ ω―–ΨΩΫαΙϊ‘Λ ΨΕ© ικΡΘ”ΘΝΘ»Θ¬Θσ ”–«±ΝΠ≥…ΈΣ÷ΈΝΤΑ©÷ΔΜρ’ΏΤδΥϊΦ≤≤ΓΒΡ“©ΈοΓΘ

Θ¬ΘαΘχ « Θ¬ΘψΘλ⁃Θ≤ Φ“ΉεΒΑΑΉ÷–“ΜΗω¥ΌΒρΆω“ρΉ”ΓΘΥϋΈΜ”ΎœΗΑϊΜυ÷ ÷–Θ§Τδ±ΜΦΛΜνΚσΜα”’ΒΦœΗΑϊΒΡΥάΆωΓΘΘ¬ΘψΘλ⁃Θ≤ Β»ΩΙΒρΆωΒΑΑΉΩ…“‘”κ Θ¬ΘαΘχ œύΜΞΉς”ΟΘ§“÷÷ΤœΗΑϊΒΡΥάΆωΓΘΡΩ«ΑΘ§ΩΤ―ßΦ““Μ÷¬»œΈΣœΗΑϊΒΡΒρΆω «Ά®ΙΐœΗΑϊΒρΆωΒΑΑΉ”κΩΙΒρΆωΒΑΑΉœύΜΞΉς”Οά¥ΒςΫΎΒΡΘ§»ΜΕχ‘ΎΒρΆω”ΠΦΛΖ¥”Π÷–ΦΛΜν Θ¬ΘαΘχ ΚΆ Θ¬ΘαΘκ ΒΡΜζάμ»‘»Μ¥φ‘Ύ’υ¬έΓΘΘ«ΘαΘωΘαΘτΘηΘιΘοΘτΘιΘσ Β»ΖΔœ÷Ε© ικΡ Θ”ΘΝΘ»Θ¬Θσ Ω…“‘÷±Ϋ”ΦΛΜν Θ¬ΘαΘχ ΒςΫΎΒΡœΏΝΘΧεΒρΆωΘέΘ¥Θ≤ΘίΓΘΆ®Ιΐ Θ¬ΘιΘμ Θ”ΘΝΘ»Θ¬”κ Θ¬ΘαΘχ Η¥ΚœΈοΒΡΕΰΈ§Θ±ΘΒΘΈ⁃Θ±Θ» “λΚΥΒΞΝΩΉ”œύΙΊΚΥ¥≈Ι≤’ώΤΉ”κΥ≥¥≈≥Ύ‘Ξ‘ω«ΩΚΥ¥≈Ι≤’ώ Β―ιΘ§ΥϊΟ«»ΖΕ® Θ¬ΘαΘχΒΡΦΛΜνΈΜΒψ”κΩΙΒρΆωΒΑΑΉΒΡΒδ–ΆΡΘ ΫΆξ»Ϊ≤ΜΆ§ΓΘΘ¬ΘαΘχ ΦΛΜνΖ¥”Π“ΐΤπΝΥ“ΜœΒΝ–Ε·Χ§Ν§–χ±δΜ·Θ§Αϋά®ΙΙœσΒΡΗΡ±δ”κΤκΨέΖ¥”ΠΓΘΆ®Ιΐ“ΜœΒΝ–Ε© ικΡ”κ Θ¬ΘΝΘαΘχΫαΚœ Β―ιΖΔœ÷Θ§÷Μ“ΣΧΦΝ¥÷ßΦή≤Μ «ΈΜ”ΎΫαΚœΈΜΒψΘ§Θ¬ΘιΘμ Θ”ΘΝΘ»Θ¬ ΨυΡή”––ßΒΊ”κ Θ¬ΘαΘχ Ής”ΟΓΘΗΟΙΛΉςΈΣΑ©÷ΔΒΡ÷ΈΝΤΧαΙ©ΝΥ“ΜΧθ–¬ΒΡΥΦ¬ΖΘ§Φ¥Ϋ®ΝΔœΗΑϊΒρΆωΒςΫΎΒΡ“ΜΗω–¬ΒΡΑ–±ξΘ§―Α’“Ε© ικΡά¥ΦΛΜνΑ©œΗΑϊ÷–ΒΡœΗΑϊΒρΆωΆ®¬ΖΓΘ
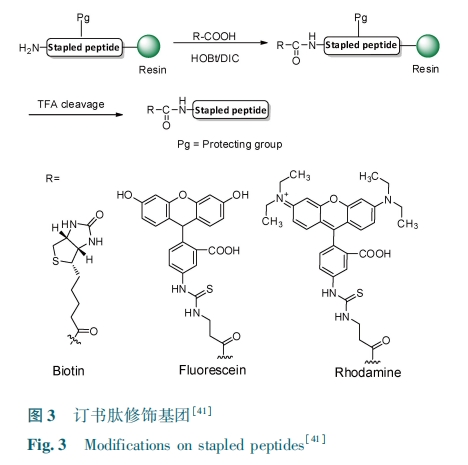
Θ¥.Θ≤ Ε© ικΡ‘ΎΑ§ΉΧ≤Γ÷ΈΝΤ÷–ΒΡ”Π”Ο
»ΥάύΟβ“Ώ»±œί≤ΓΕΨΘ®Θ»Θ…Θ÷Θ© τ”ΎΖ¥ΉΣ¬Φ≤ΓΕΨΒΡ“Μ÷÷ΓΘΘ»Θ…Θ÷ Ά®ΙΐΤΤΜΒ»ΥΧεΒΡΟβ“ΏΡήΝΠΘ§ΒΦ÷¬Οβ“ΏœΒΆ≥Ε‘ΩΙ‘≠ ß»ΞΒ÷ΩΙΝΠΘ§¥”Εχ“ΐΖΔΗς÷÷Φ≤≤ΓΑϋά®Α©÷ΔΓΘΘ»Θ…Θ÷⁃Θ± “¬Ω«ΒΑΑΉ‘Ύ≤ΓΕΨΉιΉΑ÷–Αγ―ίΉ≈÷Ί“ΣΒΡΫ«…ΪΘ§≥…ΈΣ–¬–ΆΑ§ΉΧ≤ΓΝΤΖ®ΒΡ“ΜΗω÷Ί“ΣΒΡΑ–±ξΓΘ―–ΨΩ’Ώ‘χ±®Βά“ΜΗωΚ§ Θ±Θ≤ ΗωΑ±ΜυΥαΒΡ¥χ”– ΠΝ ¬ί–ΐΫαΙΙΒΡΕύκΡΘ®ΘΟΘΝΘ…Θ© Ω…“‘‘ΎΧεΆβΑ–œρ“¬Ω«ΒΡΧΦΕΥ«χ”ρΘ® ΘΟ⁃ΘΟΘΝΘ©ΉηΕœ≤ΓΕΨ“¬Ω«ΒΡΉιΉΑΘέΘΒΘ±ΘίΓΘΒΪ «Θ§ΗΟΕύκΡ“ρ≤ΜΡήΫχ»κœΗΑϊΡΛΕχ‘Ύ…ζΈοΧεΡΎ≤ΜΡήΖΔ…ζΉς”ΟΘ§≤Μ ΚœΉςΈΣΩΙ≤ΓΕΨΒΡ“©ΈοΓΘΘΟΘΝΘ… ‘ΎΗ¥ΚœΈο ΘΟΘΝΘ…⁃ ΘΟ⁃ΘΟΘΝ ΫαΚœΒΡΫαΙΙ“―Ψ≠Ά®ΙΐΗΏΖ÷±φ ΘΊ …δœΏΨßΧε―ή…δΖ®±ΜΫβΈωΘέΘΒΘ≤ΘίΓΘΘΎΘηΘαΘνΘγ Β»ΘέΘ≥ΘΗΘί Ά®ΙΐΫαΙΙ”≈Μ·ΫΪ ΘΟΘΝΘ… ΉΣ±δ≥…Ω…“‘Ϋχ»κœΗΑϊΡΛΒΡΕ© ικΡΘ®ΘΈΘΌΘΝΘΡ⁃Θ±Θ©ΓΘ Ήœ»Θ§ΥϊΟ«άϊ”Ο‘≤Εΰ…ΪΤΉ≤β ‘»ή“Κ÷– ΘΈΘΌΘΝΘΡ⁃Θ± ”κ ΘΟΘΝΘ… ΒΡΕΰΦΕΫαΙΙΘ§ΖΔœ÷ΘΟΘΝΘ… ÷ς“Σ“‘ΈόΙφ‘ρΨμ«ζΒΡ–Έ Ϋ¥φ‘ΎΘ§≤ΔΈόΒδ–ΆΒΡ ΠΝ¬ί–ΐΫαΙΙΓΘ’β“ΜΫαΙϊΦδΫ”÷ß≥÷ΝΥΫαΚœ”’ΒΦ ΘΟΘΝΘ… ΒΡΙΙœσΖΔ…ζ±δΜ·ΒΦ÷¬ ΘΟΘΝΘ… ”κ ΘΟ⁃ΘΟΘΝ Η¥ΚœΈοΒΡ–Έ≥…ΓΘ≤ΜΆ§ΒΡ «Θ§ΘΈΘΌΘΝΘΡ⁃Θ± ”–Ή≈Οςœ‘ΒΡ ΠΝ ¬ί–ΐΫαΙΙΘ§Τδ¬ί–ΐ≥ΧΕ»¥σ‘Φ ΈΣ ΘΗΘΑΘΞ ΓΘΆ® Ιΐ ΚΥ ¥≈ ΤΉ ΆΦ ”≥ …δΘ§ Υϊ Ο« ΖΔ œ÷ΘΈΘΌΘΝΘΡ⁃Θ± ΚΆ ΘΟΘΝΘ… Ζ÷±π”κ ΘΟ⁃ΘΟΘΝ ΫαΚœΒΡΜ·―ßΈΜ“ΤΖ«≥ΘœύΥΤΘ§¥”Εχ÷ΛΟςΤδΫαΚœΖΫ Ϋ“≤Ζ«≥ΘœύΥΤΓΘΆ®ΙΐΙ≤ΨέΫΙœ‘ΈΔΨΒ Β―ιΘ§÷Λ ΒΕ© ικΡΩ…“‘Ϋχ»κœΗΑϊΡΛΘΜΆ®ΙΐΧεΆβœΗΑϊ Β―ιΘ§ΥϊΟ«÷Λ ΒΝΥ ΘΈΘΌΘΝΘΡ⁃Θ± Ω…“‘“÷÷Τ≤ΓΕΨΒΡΉιΉΑΓΘ‘Ύ≤ΓΕΨ«÷»ΨœΗΑϊ Β―ι÷–Θ§ΘΈΘΌΘΝΘΡ⁃Θ± ΟΜ”–»ΈΚΈΜν–‘Θ§±μΟςΕ© ικΡΕ‘≤ΓΕΨΫχ»κœΗΑϊΡΛΒΡΙΐ≥ΧΟΜ”–“÷÷ΤΉς”ΟΓΘ’β“≤Ϋχ“Μ≤Ϋ»ΖΕ® ΘΈΘΌΘΝΘΡ⁃Θ± Ά®Ιΐ÷±Ϋ”ΫαΚœ‘ΎΘΟΘΝ …œ”ΑœλΕΰΨέΫγΟφΒΡ–Έ≥…Θ§ΉηΑ≠≥… λΒΡΚΆΈ¥≥… λΒΡ≤ΓΕΨ “¬ Ω« ΒΡ –Έ ≥…Θ§ Φθ …Ό ≤Γ ΕΨ Η– »Ψ »Υ Χε ΡΎ œΗ ΑϊΓΘΘΈΘΌΘΝΘΡ⁃Θ± «Ε‘”ΎΩΙ Θ»Θ…Θ÷⁃Θ± ”–Ή≈ΙψΤΉΒΡΩΙ≤ΓΕΨΜν–‘Θ§”–Ή≈±Μ”≈Μ·≥…ΈΣ“Μ÷÷–¬ΒΡ÷ΈΝΤΑ§ΉΧ≤Γ“©ΈοΒΡ«±ΝΠΓΘ
–μΕύΝ¥ΕΈ≥ΛΒΡΕύκΡ”…”ΎΫαΙΙΒΡ…Ξ ßΚΆΟΗΫβΈ»Ε®–‘≤νΕχ÷¬ ΙΤδ…ζΈοάϊ”Ο–‘ΒΆΓΘΕςΗΞΈΛκΡ «”… Θ≥ΘΕ ΗωΑ±ΜυΥαΉι≥…ΒΡΕύκΡΘ§ «ΒΎ“ΜΗωΩ…“‘ΉηΕœ Θ»Θ…Θ÷⁃Θ± Ϋχ»κ»ΥΧεΒΡΡΛ»ΎΚœ“÷÷ΤΦΝΓΘΥϋΆ®ΙΐΑ–œρ≤ΓΕΨΒΡ»ΎΚœΤ§ΕΈά¥“÷÷Τ Θ»Θ…Θ÷⁃Θ± ≤ΓΕΨΒΡΗ–»ΨΓΘΒΪ «Θ§”…”Ύ…ζΈοΈ»Ε®–‘≤νΘ§≤ΜΡήΩΎΖΰΘ§ΕςΗΞΈΛκΡ“Μ÷±±Μœό÷Τ≥…ΈΣ≤ΙΨ»–ΆΒΡ“ΫΝΤ―Γ‘ώΓΘΈΣΝΥΩΥΖΰ”ΟΉς“ΫΝΤΕύκΡΒΡΟΗΫβΈ»Ε®–‘≤νΘ§Θ¬ΘιΘρΘδ Β»ΘέΘΒΘ≥Θί…ηΦΤ≤ΔΚœ≥…ΝΥΧΦΧΦΙ«ΦήΒΡΙΙœσΥχΕ®ΒΡΕςΗΞΈΛκΡΘ®»γ±μ Θ≥ Υυ ΨΘ©Θ§≤ΔΕ‘ΤδΫχ––ΝΥ…ζΈοΈοάμΚΆ…ζΈο“©άμ―ß”Αœλ―–ΨΩΓΘΥϊΟ«―Γ»ΓΕςΗΞΈΛκΡΘ®ΘιΘ§Θι ΘΪ Θ¥Θ©ΒΡΈΜ÷Ο≤ε»κΖ«Χλ»ΜΑ±ΜυΥαΘ§Ψ≠Ιΐœ©ΧΰΗ¥Ζ÷Ϋβ–Έ≥…ΙΙœσΥχΕ®ΒΡΕςΗΞΈΛκΡΓΘΗυΨί ΘγΘπΘ¥Θ± ΒΡΨßΧεΫαΙΙ―Γ‘ώΖ«Χλ»ΜΑ±ΜυΥα≤ε»κΈΜΒψ“‘»Ζ±ΘΕ© ικΡ≤ΜΜα¥ρΕœ Θ»Θ“Θ± ”κΘ»Θ“Θ≤ ÷±Ϋ”œύΜΞΉς”ΟΒΡ―œΗώΒΡ ηΥ°ΫγΟφΓΘΨ≠ΙΐΟ”ΒΑΑΉΟΗΒΡΟΗΫβΈ»Ε®–‘≤β ‘Θ§ΥϊΟ«ΖΔœ÷Κ§”–“ΜΗω÷ßΦήΒΡΕςΗΞΈΛκΡΒΡΟΗΫβΈ»Ε®‘Ε«Ω”ΎΈ¥–ό ΈΒΡΕςΗΞΈΛκΡΘΜΚ§”–ΝΫΗω÷ßΦήΒΡΕςΗΞΈΛκΡ±»“ΜΗω÷ßΦήΒΡΕςΗΞΈΛκΡ”–Ή≈Ηϋ«ΩΒΡΟΗΫβΈ»Ε®–‘ΓΘΩΙΕΨΜν–‘ Β―ι±μΟςΚ§”–ΝΫΗω÷ßΦήΒΡΕςΗΞΈΛκΡΩ…“‘”––ßΒΊ“÷÷Τ≤ΓΕΨΒΡΗ–»ΨΓΘ¥ΥΆβΘ§–Γ σΧεΡΎΟΗΫβΈ»Ε®–‘ Β―ιΖΔœ÷ΨΏ”–ΝΫΗω÷ßΦήΒΡΕ© ικΡ‘ΎΧεΡΎΒΡΑκΥΞΤΎ”–Ή≈œ‘÷χΒΊΧαΗΏΘ§Ο÷≤ΙΝΥΕύκΡάύΘ»Θ…Θ÷⁃Θ± ΡΛ»ΎΚœ“÷÷ΤΦΝΒΡ“©¥ζΕ·ΝΠ―ß»±œίΓΘΨ≠Ιΐ–Γ σ Β―ιΖΔœ÷¥ρΝΫΗω÷ßΦήΒΡΕςΗΞΈΛκΡ”–“ΜΕ®ΒΡΩΎΖΰάϊ”ΟΕ»Θ§‘ΎΝι≥ΛάύΕ·Έο÷–Τδ«±‘ΎΒΡΩΎΖΰάϊ”ΟΕ»”–¥ΐΫχ“Μ≤ΫΒΡ―–ΨΩΓΘ

Θ¥.Θ≥ Ε© ικΡ‘Ύ–≈Κ≈Ά®¬Ζ÷–ΒΡ”Π”Ο
Θ¥.Θ≥.Θ± ΒςΩΊ ΘΈΘοΘτΘψΘη –≈Κ≈
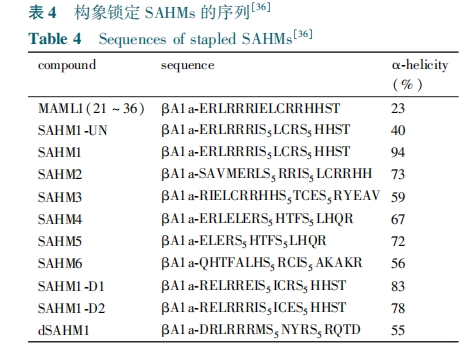
ΘΈΘοΘτΘψΘη –≈Κ≈Ά®¬Ζ «”…Ψ÷≤ΩœΗΑϊΦδœύΜΞΉς”ΟΕχ≤ζ…ζΒΡΘ§≤Δ”κ÷ΉΝω–Έ≥…ΚΆΡ≥–©…ώΨ≠œΒΆ≥Φ≤≤Γ”–Ή≈Οή«–ΒΡΙΊœΒΘ§ΨΏ”–Η¥‘”Εύ―υΒΡΙΠΡήΘ§»γ≤Έ”κ‘λ―ΣΓΔΘ‘ œΗΑϊΖΔ”ΐΚΆ―ΣΙή…ζ≥…Β»…ζάμΙΐ≥ΧΓΘΘΈΘοΘτΘψΘη ΒΑΑΉ≤Έ”κ“ΜΗω±Θ ΊΒΡ–≈Κ≈Ά®¬ΖΘ§Ω…“‘ΒςΫΎœΗΑϊΒΡΖ÷Μ·ΓΔ‘ω÷≥ΚΆΥάΆωΓΘΘΈΘοΘτΘψΘη –≈Κ≈ΒΡ≥÷–χ ±ΦδΗζ«ΩΕ»±Μ―œΗώΒςΩΊΘ§‘Ύ–μΕύΦ≤≤Γ÷– ΘΈΘοΘτΘψΘη ΒΑΑΉΙΠΡή…Ξ ßΒΡΆΜ±δ“―Ψ≠±ΜΖΔœ÷ΓΘΘΈΘοΘτΘψΘη ΙΠΡήΜώΒΟ–‘ΒΡΆΜ±δ”κΑ©÷ΔΒΡΖΔ…ζ”–ΙΊΓΘ‘ΎΦ±–‘ Θ‘ ΝήΑΆœΗΑϊΑΉ―Σ≤Γ±μ¥οΒΡ ΘΆΘΝΘΆΘΧΘ± ΒΡœ‘–‘“÷÷ΤΥιΤ§Θ® ΘδΘνΘΆΘΝΘΆΘΧΘ±Θ© ±μΟς”–Ή≈“÷÷Τ ΘΈΘοΘτΘψΘη –≈Κ≈ΚΆœΗΑϊ‘ω÷≥ΒΡΡήΝΠΓΘ“ΜΗωΙΙœσΥχΕ®ΒΡ ΘδΘνΘΆΘΝΘΆΘΧΘ± ΥιΤ§Θ§Ω…Ρή“÷÷Τ»Ϊ≥ΛΒΡ ΘΆΘΝΘΆΘΧΘ± ”κ Θ…ΘΟΘΈΘ±⁃ΘΟΘ”ΘΧ Η¥ΚœΈοΒΡΫαΚœΓΘ«χ±π”ΎΈ¥–ό ΈΒΡΕύκΡΘ§Ε© ικΡ”–Ή≈ΗϋΗΏΒΡΫαΚœΑ–œρΒΑΑΉΡήΝΠΘ§ΗϋΗΏΒΡ¥ζ–ΜΈ»Ε®–‘ΚΆ―ΣΫ§ΑκΥΞΤΎΓΘ
ΘΆΘοΘεΘλΘλΘεΘρΘιΘνΘγ Β»ΘέΘ≥ΘΕΘίΗυΨί»Υάύ ΘΈΘοΘτΘψΘηΘ± ΥΡΨέΧεΗ¥ΚœΈοΒΡΫαΙΙ…ηΦΤΝΥ“ΜœΒΝ–ΙΙœσΥχΕ®ΒΡ ΠΝ ¬ί–ΐΒΡΕύκΡΘ®Θ”ΘΝΘ»ΘΆΘσΘ©Θ§»γ±μ Θ¥ Υυ ΨΓΘΆ®Ιΐ≤β ‘Ε© ικΡΒΡ ΠΝ ¬ί–ΐ≥ΧΕ»ΓΔ»κΡΛ–‘ΓΔ”κ Θ…ΘΟΘΈΘ±⁃ΘΟΘ”ΘΧ Η¥ΚœΈο”κ ΘΆΘΝΘΆΘΧΘ± ΨΚ’υΫαΚœΡήΝΠΘ§ΥϊΟ«…Η―Γ≥ωάμœκΒΡΕ© ικΡ Θ”ΘΝΘ»ΘΆΘ±ΓΘΆ®ΙΐΖ÷Έω ΘΈΘοΘτΘψΘηΘ± –≈Κ≈Α–œρΜυ“ρΒΡΥ°ΤΫΚΆ Θ”ΘΝΘ»ΘΆΘ± ”’ΒΦΒΡ»Ϊ±μ¥οΤΉΘ§ΥϊΟ«÷Λ ΒΝΥ Θ”ΘΝΘ»ΘΆΘ± Ω…“‘ΧΊ“λ–‘“÷÷Τ»ΥΧε”κ–Γ σΒΡ ΘΈΘοΘτΘψΘη –≈Κ≈Ά®¬ΖΘ§¥”ΕχΉηΑ≠Φ±–‘ Θ‘ ΝήΑΆœΗΑϊΑΉ―Σ≤ΓœΗΑϊΒΡ…ζ≥…ΓΘΗΟΙΛΉςΕ‘ Θ”ΘΝΘ»ΘΆΘ± ¥φ‘Ύ«±‘ΎΒΡ“ΫΝΤΦέ÷ΒΧαΙ©άμ¬έΜυ¥ΓΘΜΘ”ΘΝΘ»ΘΆΘ± «ΖώΩ…“‘≥…ΈΣ“ΫΝΤ“©Έο»‘–ηœΒΆ≥ΒΡ―–ΨΩΓΘ
Θ¥.Θ≥.Θ≤ ΒςΩΊ ΘΉΘνΘτ –≈Κ≈
ΘΉΘνΘτ –≈Κ≈Ά®¬ΖΩ…“‘ΒςΩΊœΗΑϊΒΡ–ΈΧ§ΓΔ‘ΥΕ·ΓΔ‘ω÷≥ΚΆΖ÷Μ·ΓΘ‘Ύ≈ΏΧΞΖΔ”ΐΓΔ≥…»ΥΉι÷·ΚψΕ®ΚΆΉι÷·‘Ό…ζ÷–Θ§ΘΉΘνΘτ –≈Κ≈Ά®¬ΖΑγ―ίΉ≈÷Ί“ΣΒΡΫ«…ΪΓΘ“λ≥ΘΒΡ ΘΉΘνΘτ –≈Κ≈Ά®¬ΖΒΡΜνΜ·”κ¥σΕύ ΐΒΡ÷ΉΝωΖΔ…ζ”–ΙΊΘ§»ΜΕχΗΟ–≈Κ≈Ά®¬ΖΒΡ“÷÷Τ“≤ΜαΒΦ÷¬–μΕύΦ≤≤ΓΘ§άΐ»γΘ§Ι«÷ ηΥ…÷ΔΓΔ…ώΨ≠ΆΥ–––‘Φ≤≤ΓΓΔΧ«Ρρ≤ΓΚΆ÷λ≤°ΧΊΉέΚœ÷ΔΓΘΦΛΜνΜρ’Ώ“÷÷Τ ΘΉΘνΘτ –≈Κ≈Ά®¬ΖΩ…ΡήΧαΙ©“ΜΗω”–”ΟΒΡΙΛΨΏ»ΞΝΥΫβΗ…œΗΑϊΒΡΉ‘Έ“Ηϋ–¬ΓΔΖ÷Μ·ΚΆΉι÷·ΒΡ–Έ≥…ΓΘΘΝΘχΘιΘν÷±Ϋ””κ Π¬ Ν§ΜΖΒΑΑΉΉς”ΟΘ§Χ«‘≠ΚœΟΗΦΛΟΗ Θ≥Π¬ ΒςΩΊΘΝΘχΘιΘν ΒΡΝΉΥαΜ·ΚΆΖΚΥΊΜ·Θ§»γΆΦ ΘΒ Υυ ΨΓΘ
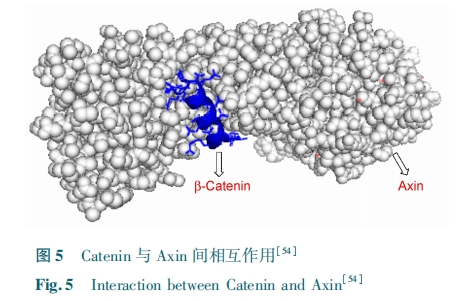
ΉνΫϋΘ§ΘΟΘθΘι Β»ΘέΘΒΘ¥Θί…ηΦΤΝΥΩ…“‘ΦΛΜν ΘΉΘνΘτ –≈Κ≈Ά®¬ΖΒΡΡή»κΡΛΒΡΕ© ικΡΓΘΗΟΕύκΡ÷Φ‘Ύ¥ρΤΤ ΘΝΘχΘιΘν ”κ Π¬⁃Ν§ΜΖΒΑΑΉ÷°ΦδΒΡΉς”ΟΓΘΥϊΟ«Μ·―ßΚœ≥…ΝΥΝΫΧθΕ© ικΡΘ”ΘΝΘ»Θ–ΘΝΘ± ”κ Θ”ΘΝΘ»Θ–ΘΝΘ≤ΘΜΆ®Ιΐ‘≤Εΰ…ΪΤΉ Β―ιΖΔœ÷Θ§”κœΏ–‘ΒΡΕύκΡ ΘΝΘχΘιΘν œύ±»Θ§ΝΫΧθΚœ≥…ΒΡΕ© ικΡΒΡ ΠΝ ¬ί–ΐ≥ΧΕ»Ος œ‘ Χα ΗΏΓΘΧε Άβ ΘπΘθΘλΘλ⁃ΘδΘοΘςΘν Β ―ι ΐ Ψί ±μ ΟςΘ”ΘΝΘ»Θ–ΘΝΘ± ΫαΚœ Π¬ Ν§ΜΖΒΑΑΉΒΡΫαΚœΡήΝΠ±» Θ”ΘΝΘ»Θ–ΘΝΘ≤ Ηϋ«ΩΓΘœΗ Αϊ »κ ΡΛ –‘ Β ―ι ΚΆ Οβ “Ώ Ι≤ ≥Ν Βμ Β ―ι ±μ ΟςΘ”ΘΝΘ»Θ–ΘΝΘ± Ά®Ιΐ”κ Π¬⁃Ν§ΜΖΒΑΑΉΒΡΫαΚœΩ…“‘”––ßΒΊ¥ρΤΤΡΎ‘¥ΒΡ ΘΝΘχΘιΘν⁃Π¬⁃Ν§ΜΖΒΑΑΉΗ¥ΚœΈοΒΡ…ζ≥…ΓΘΥϊΟ«ΧΫΥςΝΥΕ‘ΉΣ¬ΦΜν–‘ΒΡ”ΑœλΘ§±μΟς Θ”ΘΝΘ»Θ–ΘΝΘ± Ω…“‘ΗΏΕ»ΧΊ“λ–‘ΒΊΦΛΜν ΘΉΘνΘτ –≈Κ≈ΓΘ
Θ¥.Θ¥ Ε© ικΡ‘ΎΗΈ―Ή÷ΈΝΤ÷–ΒΡ”Π”Ο
±ϊ–ΆΗΈ―Ή «“Μ÷÷»Ϊ«ρΝς–––‘¥Ϊ»Ψ≤ΓΘ§Τδ÷ς“Σ≤Γ‘≠Χε±ϊΗΈ≤ΓΕΨΘ®Θ»ΘΟΘ÷Θ© τ”ΎΜΤ≤ΓΕΨΦ“ΉεΘ§ «ΨΏ”–Φ‘ΡΛΒΞΙ…’ΐΝ¥ΒΡ Θ“ΘΈΘΝ ≤ΓΕΨΓΘΘΟΘΡΘΗΘ± « Θ»ΘΟΘ÷ Ϋχ»κΥό÷ςœΗΑϊΒΡ÷Ί“Σ ήΧεΘ§“ρ¥Υ»γΚΈΉηΕœ±ϊΗΈ≤ΓΕΨ”κ ΘΟΘΡΘΗΘ± ΒΡœύΜΞΉς”Ο≥…ΈΣ―–ΖΔ±ϊΗΈ≤ΓΕΨ»κΡΛ“÷÷ΤΒΡ÷Ί“ΣΑ–±ξΓΘΘΟΘθΘι Β»ΘέΘΒΘ≥ΘίΫΪΙΙœσΥχΕ®≤Ώ¬‘”Ο”Ύ…ηΦΤ Θ»ΘΟΘ÷ »κΡΛ“÷÷ΤΘ§ΥϊΟ«Κœ≥…ΝΥ“ΜœΒΝ–Ω…“‘ΡΘΡβ ΘΟΘΡΘΗΘ± ΒΡ ΘΧΘ≈ΘΧ⁃ΘηΘεΘλΘιΘχ ΘΡ«χΒΡΕύκΡΘ§ΕύκΡΫαΙΙ»γ±μ ΘΒ Υυ ΨΓΘΆ®Ιΐ≤β ‘’β–©Ε© ικΡΩΙ Θ»ΘΟΘ÷ Η–»ΨΜν–‘Θ§ΥϊΟ«ΖΔœ÷ Θ”ΘΝΘ»Θ»ΘΒ Ε‘ Θ»ΘΟΘ÷ Η–»Ψ”– Ϋœ ΗΏ ΒΡ “÷ ÷Τ Μν –‘ΓΘ‘Ύ Θ»ΘΟΘ÷ΘψΘψ⁃Θ ΘΤΘ»Θ±ΓΔ Θ»ΘΟΘ÷ΘπΘπ⁃Θ ΘΤΘ»⁃Θ± ΚΆ Θ»ΘΟΘ÷ΘπΘπ⁃ΘψΘοΘνΘ± ΧεœΒ÷–Θ§Θ”ΘΝΘ»Θ»⁃ΘΒ Ε‘ Θ»ΘΟΘ÷ ΒΡΑκ ΐ“÷÷Τ≈®Ε»Ζ÷±πΈΣ Θ≥ΘΙΓΔΘ±ΘΖ ΚΆ Θ≥ΘΒ ΠΧΘΆΓΘΥϊΟ«≤β ‘ΝΥΕ© ικΡΒΡΩΙΒΑΑΉΟΗΫβΡήΝΠΘ§ΖΔœ÷Ε© ικΡΩΙΒΡΒΑΑΉΟΗΫβΡήΝΠΟςœ‘ΗΏ”ΎœΏ–‘ΒΡΈ¥–ό ΈΕύκΡΓΘΗΟΙΛΉς « Ή¥ΈΫΪΙΙœσΥχΕ®≤Ώ¬‘”Π”ΟΒΫ Θ»ΘΟΘ÷ »κΡΛ“÷÷ΤΦΝΝλ”ρΘ§«“≥…ΙΠ’“ΒΫΗΏΜν–‘ΒΡΕ© ικΡ Θ”ΘΝΘ»Θ»⁃ΘΒΘ§ΈΣ Θ»ΘΟΘ÷ ΒΡ÷ΈΝΤΧαΙ©ΝΥ–¬ΒΡΖ÷Ή”ΙΛΨΏΓΘ
ΘΒ Ε© ικΡΒΡ«ΑΨΑ”κ’ΙΆϊ
Θ÷ΘεΘρΘδΘιΘνΘε Β» Ή¥ΈΖΔΟς≥ωΧΦΧΦΝ¥÷ßΦήΒΡΕ© ικΡ÷ΝΫώΘ§¥”Κœ≥…ΖΫΖ®ΚΆ…ζΈο”Π”ΟΝΫΖΫΟφΘ§ΨυΒΟΒΫΝΥΩλΥΌΒΡΖΔ’ΙΓΘ¥ΪΆ≥–ΓΖ÷Ή”ΧεΜΐΙΐ–Γ≤ΜΡή”––ßΒΊΒςΫΎΒΑΑΉΒΑΑΉœύΜΞΉς”ΟΓΘΕ© ικΡ≤ΜΫωΩ…“‘ΉςΈΣ“ΜΗω«±‘ΎΒΡ“ΫΝΤΑ–œρΘ§Εχ«“Ω…“‘ΉςΈΣ“ΜΗω–¬ΒΡΙΛΨΏ”Ο”Ύ Ε±π–¬ΒΡΖ¥”ΠΈΜΒψΦΑ–¬ΒΡ≈δΧεΒ»ΓΘΕ© ικΡ±»–ΓΖ÷Ή””–ΗϋΕύΒΡ―Γ‘ώ–‘Θ§ΤδΦέΗώ”÷‘ΕΒΆ”ΎΩΙΧεΒ»…ζΈο÷ΤΦΝΘ§Εχ«“Ω…“‘»κΡΛΓΘΆ§―υΨΏ”–Έ»Ε®Ω’Φδ’έΒΰΫαΙΙΜΙ”––ΓΒΑΑΉΘ®”»Τδ «ΜΖΒΑΑΉΘ©ΘέΘΒΘΕ ΓΪ ΘΕΘΒΘίΘ§’β–©Ζ÷Ή”ΒΡΕάΧΊ–‘÷ Θ§ ΙΒΟΥϋΟ«≥…ΈΣΈ¥ά¥“©ΈοΖΔ’Ι÷ΒΒΟΧΫΥςΒΡ“ΜΗωΖΫœρΓΘΥφΉ≈ΕύκΡΝ§Ϋ”ΦΦ θΒΡΖΔ’ΙΘ§¥σ–ΆΕ© ικΡΒΡΚœ≥…≥…ΈΣœ÷ ΒΓΘ’βΫΪΈΣΖΔ’Ι–¬“Μ¥ζΒΑΑΉάύ“©ΈοΧαΙ©«Ω”–ΝΠΒΡΙΛΨΏΓΘΈ“Ο«”–άμ”…œύ–≈‘ΎΈ¥ά¥ΦΗΡξΕ© ικΡάύΒΡ“©Έο“‘ΦΑ–ΓΒΑΑΉ“©ΈοΜα≤ΜΕœΒΊΆΕ»κΒΫΝΌ¥≤Θ§≤ΔΜώΒΟ≥ΛΉψΒΡΖΔ’ΙΓΘ
Οβ‘π…υΟςΘΚ±ΨΈΡΈΣ––“ΒΫΜΝς―ßœΑΘ§Αφ»®Ιι‘≠Ής’ΏΦΑ‘≠‘”÷ΨΥυ”–Θ§»γ”–«÷»®Θ§Ω…ΝΣœΒ…Ψ≥ΐΓΘΈΡ’¬±ξΉΔ”–Ής’ΏΦΑΈΡ’¬≥ω¥ΠΘ§»γ–η‘ΡΕΝ‘≠ΈΡΦΑ≤ΈΩΦΈΡœΉΘ§Ω…‘ΡΕΝ‘≠‘”÷ΨΓΘ
