
摘 要 综述了多肽定量构效关系和计算机辅助多肽分子设计方法的最新进展 ,重点介绍了多肽定量构效关系研究中的化学结构定量描述符和建立数学模型的统计方法 ,并对模拟肽学和虚拟组合多肽库在多肽分子设计中的应用进行了简要的论述。
多肽是维持生命过程中必不可少的物质 ,由于它们具有高活性、高选择性以及副作用小等特点 ,现已逐渐成为药物研究的热点之一。对肽类药物的研究开发及先导化合物的发现 ,至今仍是一件耗资巨大但效率很低的工作。造成这种状况的一个主要原因就是缺乏深入的理论指导和先进的分子设计方法 ,因此迫切需要新的理论方法和多肽分子设计技术的出现。近年来 ,以各种理论计算方法和分子模拟技术为基础的计算机辅助分子设计 ,在各种肽类化合物的研究开发中得到了广泛的应用。利用计算机分子图形学、分子动力学和量子化学等进行构象分析 ,寻找多肽及类似物的药效团 ,进行二维和三维的定量构效关系(QSAR) 研究 ,及应用各种分子设计方法 ,设计有较高活性的肽类和非肽模拟物 ,已成为国际上十分活跃的研究领域。
一、多肽的定量构效关系研究
在多肽类似物的研究和开发中 ,定量构效关系是一个重要的理论计算方法和常用手段。所谓多肽的QSAR ,就是用数学模式来表达多肽类似物的化学结构信息与特定的生物活性强度间的相互关系。多肽的 QSAR 研究方法同其它药物的 QSAR 研究方法一样 ,基本上可分为以下 5 个步骤[1 ]: (1) 选择和设计一系列多肽类似物 ; (2) 类似物的化学结构的定量描述 ; (3) 合成设计化合物并进行生物活性的测定 ; (4) 建立数学模型 ,确定化学结构与生物活性之间的函数关系 ; (5) 新类似物的活性预测以及新的高活性类似物的设计。近年来 ,多肽的 QSAR 研究主要集中在如下两个方面 :多肽的化学结构定量描述符的研究和建立 QSAR 数学模型的统计方法。
在多肽的定量构效关系研究中 ,因为肽的复杂性和高柔性 ,使其以整个多肽分子为基础的参数很难确定。因此在多肽的各种 QSAR 模型中 ,普遍采用氨基酸的结构描述参数去定量描述多肽的化学结构和氨基酸残基 ,这是多肽 QSAR 分析中的一个关键问题。其基本思路是以多肽的最基本的结构信息―――氨基酸序列为基础 ,对一系列多肽类似物中变化的氨基酸残基进行定量描述 ,并把氨基酸序列转换成结构描述符矩阵的一个向量。
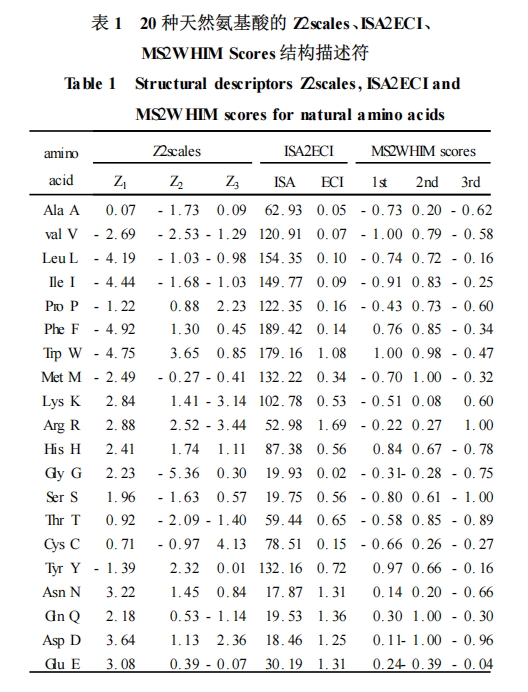
最初应用此方法的是 Kidera 等人[2 ],他们对文献报道的描述氨基酸性质的 188 个参数进行主成分分析 ,得出 10 个正交因子 ,对天然氨基酸的结构进行了定量描述。随后 ,Hellerg 等人[1 ]提出了 20 个天然氨基酸的描述符 Z2scales ,两年后 ,他们又提出了扩展的 Z2scales[3 ],成功地实现了对 35 个非天然氨基酸的定量描述。此外 ,Cocchi 等人[4 ] 用主成分分析法对用理论计算得到的内部能量进行分析 ,也获得了类似的结果 ,得到氨基酸结构描述符 t2scales。随后不久 ,Collantes 等人[5 ] 计算出了 2 个 3D 氨基酸侧链描述符 ISA (isotropic surface area) 、ECI(electroniccharge index) ;Zaliani 等人[6 ]提出的天然氨基酸的 3D结构描述符 MS2WHIM scores ,它们均来自于天然和非天然氨基酸的优化的三维结构。最近 ,又出现一种通过理论计算得出的疏水参数 SFED(solvation freeenergy density) 模型作为氨基酸的结构描述参数 ,来定量描述多肽的化学结构[7 ] 。在多肽的 QSAR 研究中 ,应用较多的氨基酸结构描述符为 Z2scales、ISA2ECI 和 MS2WHIM scores。
对一系列多肽类化合物 ,每个变化的氨基酸残基分别用 3 个 Z 值进行定量描述 ,从而实现氨基酸残基的多位置多变量的定量结构描述。但是 , Z2scales 氨基酸结构描述符并没有明确考虑到多肽类似物的构象信息 ,因此用 Z2scales 描述符来建立多肽的 QSAR 模型时 ,多肽类似物应是如下三种情况之一 : (1) 在多肽类似物中构象变化不是很大 ; (2)在用 Z2scales 进行定量结构描述时 ,已包含了一部分多肽分子的构象信息 ; (3) 建立 QSAR 模型的多肽类似物都采用了低能构象。
Collantes 等人[5 ]应用分子模拟软件搭建氨基酸的初始结构 ,而后进行构象分析、能量优化和量子化学计算等得出氨基酸的 ISA、ECI 值 ,20 种天然氨基酸的 ISA、ECI 值见表 1。ISA 近似地表示侧链基团的疏水性 ,从表 1 中的 ISA 值可见 ,它又与氨基酸分子的体积有关。ECI 表示的是氨基酸侧链基团的极性 ,即氨基酸侧链每个原子 i 的电荷数 q 的绝对值之和 ,ECI =Σ| qi| ,ECI 值能显示出侧链和受体之间的偶极作用。在一系列多肽类似物中 ,每一个氨基酸的结构信息都能根据氨基酸的序列用 ISA 和 ECI值进行定量描述。
氨基酸结构描述符 ISA2ECI 的优点是描述符个数少、来源简单 ,且它们的计算值均是来自氨基酸的三维结构 ,这样易于对多肽的 QSAR 模型进行解释分析。ISA2ECI 与 Z2scales 结构描述符相比 ,有更大的自由度 ,更易于扩展到非天然的氨基酸和肽的拟似物。如 Elizabeth 等人已成功地应用 ISA2ECI 描述符 ,对含有非天然氨基酸的多肽催产素拮抗剂进行了 QSAR 的分析。
Zaliani 等人[6 ] 在计算机上模建 20 种天然氨基酸分子 ,并进行能量优化和量化计算等 ,得出各分子的 AM1 电荷和 Connolly 表面的分子静电势 ,计算出36 个氨基酸结构描述参数。再对 20 种天然氨基酸形成的 20 行、36 列的 MS2WHIS 描述符矩阵进行主成分分析 ,从中提取出前 3 个主要成分 ,得到了 MS2WHIM scores 氨基酸结构描述符(见表 1) 。第一个指数与静电势有关 ,能体现出氨基酸残基电荷的正负性和侧链的芳香族P脂肪族的结构信息 ;第二个指数受负静电势的影响 ,并能把酸性氨基酸 Asp 和 Glu同其它氨基酸区分开 ;第三个指数多数是负值 ,但Arg 和 Lys 为正值 ,主要是和它们的正电性和线性的侧链有关。
近来 ,在多肽的 QSAR 分析中 ,也出现了不用氨基酸残基描述符来定量描述多肽化学结构的例子。如MHDV[11 ] (molecular holographic distance vector) 描述符 ,它只依靠 2D 拓扑距离矩阵和分子中的非氢原子类型 ,不需要相关的 3D 结构信息、物化性质和分子重叠 ,使用起来简单快速。但是由于缺乏一些非氢原子的原子类型 ,计算的 MHDV 描述符最多只能有 91 个 ,因此目前只能成功地进行二肽的 QSAR 的分析研究。
随着实验技术水平的提高和计算机辅助分子设计在药物分子设计中的应用 ,多肽的结构定量描述符也越来越多样化。如以上描述的由实验测定的氨基酸结构描述符 Z2scales ,及通过理论计算的指数ISA2ECI、MS2WHIM 和 MHDV , 还有其他的参数如MEDV[12 ] (molecular electronegativity2distance vector) 和指示 变 量 I[13 ], 及 多 肽 的 两 面 角 Ψ、Φ、χ1 χ2χ3[14 ,15 ]等都已成功地定量描述了多肽类似物的结构。但是 ,由于多肽分子的复杂性及较大的柔性 ,怎样进一步校正这些参数使它们更适应多肽分子的特点 ,以及怎样准确全面地定量描述多肽的三维结构信息和其它含非标准氨基酸的多肽类似物 ,仍是一个任重道远的过程。
在定量构效关系模型中 ,常用线性回归、偏最小二乘法 (PLS) 、遗传算法 ( GA) 和人工神经网络(ANN) 等统计方法 ,建立化合物的定量结构参数与生物活性之间的函数关系。通常线性回归数据分析法要求样品的数目至少 3 倍于变量的数目 ,结果才相对可靠 ,因此它不适合多肽类似物的化合物数目少、定量结构描述参数多的特点。目前 ,常用 PLS、GA 和 ANN 统计方法建立多肽的 QSAR 模型。
PLS 是在主成分分析的基础上发展起来的一种统计方法。它与主成分分析的不同在于 ,主成分分析仅仅考虑了自变量的相互作用 ,而 PLS 在考虑自变量的同时也考虑了因变量的作用 ,通过折衷各自空间内的因子 ,使模型较好地同时描述自变量和因变量。因此它能很好地克服当多肽的结构描述参数远远超过化合物的数目时 ,一般多元线性回归统计方法不能使用的缺陷。但是当结构描述符的信息比较分散时 ,用 PLS 统计方法将可能产生权重分散的问题 ,即在线性组合过程中 ,很容易将那些原始参数中占很少比例 ,但它们的累计信息对建立 QSAR 模型却很重要的一些参数忽略掉 ,以至于在以后分析QSAR 模型和分子设计中没有考虑到这些重要信息。
近年来 ,人们把遗传算法用于数据分析[16 ―21 ],发现它在定量构效关系研究中是一种很有潜力的计算工具 ,尤其在寻找最佳构效关系模型时[21 ] 。遗传算法是一类借鉴生物界自然选择和遗传机制 ,高度并行、随机、自适应的搜寻算法 ,于 20 世纪 60 年代由 Holland[22 ]创建。应用遗传算法建立多肽的 QSAR模型的优点是 : (1) 它能建立多个 QSAR 模型 ,且每个模型均能在不同侧面反映结构与活性之间的关系 ;(2) 在多数情况下 ,共线性的参数不能同时出现在一个模型中 ,因此它还能有效地避免权重分散的问题 ,而且进一步使得所建立的 QSAR 模型易于解释分析 ; (3) 遗传算法可以采用线性多项式、高次多项式和其他非线性函数等形式建立 QSAR 模型 ,因此使得多肽的 QSAR 方程更加灵活多样。目前 ,遗传算法越来越广泛地应用到各种化合物的 QSAR 分析中。遗传算法虽然比较简单 ,但很灵活 ,在不同的应用中可以存在不同的实现方法。为了发扬遗传算法的优点 ,避免它的缺点 ,遗传算法还常和其它统计方法如 GA2PLS[23 ―25 ] 、GARGS[26 ]等结合去解决问题。
人工神经网络是对生物神经网络的模拟和抽象 ,是一种由大量基本信息处理单元通过复杂的连接而成的非线性动力学系统 ,具有并行性、容错性、非线性和自学习性等特点。人工神经网络作为一种新的统计分析方法 ,现已被广泛地应用于 QSAR 的研究中[27 ―32 ] 。为了加强人工神经网络的统计分析功能 ,最近又出现了一些新的神经网络统计方法 ,如模糊人工神经网络[33 ] 、概然论神经网络(probabilisticneural networks ,PNN) 和无显著衰变神经网络 (gener2alized regression neural networks ,GRNN)[34 ]等。它们不但具有强大的学习功能 ,而且能够为 QSAR 模型提供易于解释的规则。从人工神经网络算法强大的应用潜力中可以看出 ,它是多肽的 QSAR 研究中很有前途的统计方法 ,还有待我们进一步去发掘、探索。
二、计算机辅助多肽药物分子设计方法
近 20 年来 ,科学家们发现了许多天然存在的内源性的生物活性肽 ,这些生物活性肽和受体分子的相互作用控制着特定的生理学事件 ,使得生物活性肽有可能作为治疗药物 ,且显示出了很大的优势。但肽类化合物本身作为药物有许多缺点 ,如不稳定性、生物利用度低、不易得到等。为了克服上述种种不利的因素 ,对多肽从结构上进行改造或结构替换 ,这样就形成了模拟肽学[35 ,36 ] 。模拟肽学已成为多肽药物分子设计的主要途径。目前 ,随着计算机辅助药物设计及组合化学的发展 ,虚拟组合化学库在多肽类似物的分子设计中也得到了蓬勃的发展。以下就简要的介绍模拟肽学和虚拟组合多肽库在多肽分子设计中的应用。
1. 分子设计中的模拟肽学
模拟肽学就是通过多种途径对肽从结构上进行改造或结构替换。目前改变肽结构的基本方法主要有两大类 :一类是在原有肽骨架 (主链) 基础上进行改造 ,主要的途径有环肽的分子设计方法及受约束的氨基酸替换的方法 ;另一类是非肽结构模拟。用这两类结构改造的分子都避免了母体肽分子的高柔性 ,且分子构象采取所需的构象 ,这样设计出来的药物候选物可以把代谢稳定性和口服生物有效性结合起来。
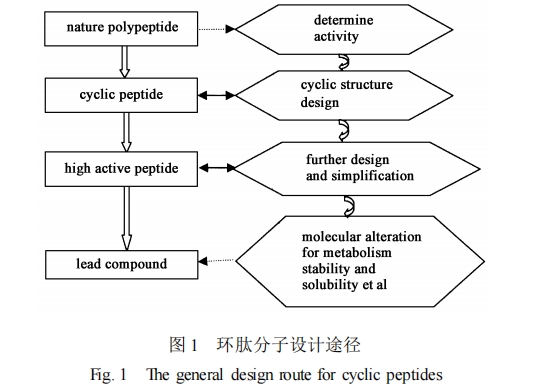
受约束(或化学修饰) 氨基酸替换的方法是 ,在原有肽骨架的基础上 ,把约束氨基酸引入生物活性肽的氨基酸序列 ,限制两面角 Φ、Ψ 和ω的旋转 ,形成局部的约束 ,并约束由共价或非共价立体相互作用所决定的侧链构象 ,进而改变生物活性肽的稳定性和活性。这些约束氨基酸可作为构象探针 ,用来探求和解释是何种局部构象决定一种特定肽的生物活性。经典的构象探针是丙氨酸 ,用丙氨酸来逐个替换肽链中的每个氨基酸 ,在尽可能保持构象不变的情况下 ,决定每个氨基酸对活性的影响。在其他氨基酸模型中 ,D 型氨基酸是最常用的残基 ,而且常常起关键作用 ,如在肽链的酶解点引入 D 型氨基酸 ,可以提高肽链的抗酶解能力和活性。近年来 ,关于约 束 氨 基 酸 的 例 子 很 多 , 如 α2甲 基 化 氨 基酸[38 ,39 ] 、Nα2Cα 环化氨基酸[40 ,41 ] 和 α2烷基环烷羧酸[42 ,43 ]等。可见 ,用天然的或非天然的氨基酸进行取代分子设计 ,对侧链进行优化 ,可以提高化合物的活性及其与受体的选择性 ,进而设计出高活性的化合物。
非肽结构模拟就是替换肽骨架 ,同时保留其药效团及其所需的三维排布 ,设计新的非肽小分子。进行非肽结构模拟的前提是确定生物活性肽的三维药效团 ,所谓三维药效团就是生物活性肽与受体结合(或作用) 时 ,起重要作用的原子或基团的空间距离限制及它们的空间三维结构排布。一个成功的三维药效团模型 ,包含了药物与受体结合部位的一些结构信息 ,此信息可用来指导分子设计。因此 ,如果设计的非肽小分子符合三维药效团模型的要求 ,它就可能有活性。构建生物活性肽的三维药效团的一般步骤为[37 ]: (1) 选择一系列与受体亲和性高的肽类似物组成一个分子集合 ,从这些分子中选出药效基团元素 ,在所有的肽类似物分子中 ,药效基团元素的数目必须相同 ,分子刚性要大 ,而且所测定的生物活性数据应有很好的同源性 ; (2) 对多肽类似物集合中的每一个活性化合物进行构象分析 ,得到它们的低能构象 ; (3) 用分子叠合方法选择高活性肽类似物的活性构象 ,分子叠合时用药效基团元素作为分子间原子叠合对 ,以此建立化合物的三维药效团。生物活性肽的三维药效团模型确定后 ,保持其三维药效团 ,通过替换肽骨架来进行非肽小分子设计。通常设计的小分子化合物能克服肽类似物普遍存在的生物利用率低、代谢速度快、不适于口服等限制 ,可见非肽结构模拟是鉴定和优化潜在肽类药物的可行的途径之一。
在模拟肽学的分子设计中应该注意以下几个关键问题[35 ,36 ]:在第一代的模拟肽中保持一定的构象柔性 ;保持肽侧链的药效团 ;用尽量多的非肽结构置换肽骨架 ;肽类药物的靶标应有现成的假定药效基团模型。
2. 虚拟组合多肽库
近年来 ,组合化学和高通量筛选技术的迅速发展 ,对传统的先导化合物的发现和优化产生了巨大的影响。特别是随着计算化学与分子模拟技术的发展 ,计算机构建虚拟样品库的方法已用于药物设计 ,并在先导化合物的研究和开发中起了重要的作用[44 ―46 ] 。与此同时 ,虚拟组合多肽库也在肽类药物的设计中得到了应用[47 ] 。以下举例说明虚拟组合多肽库的构建及应用。
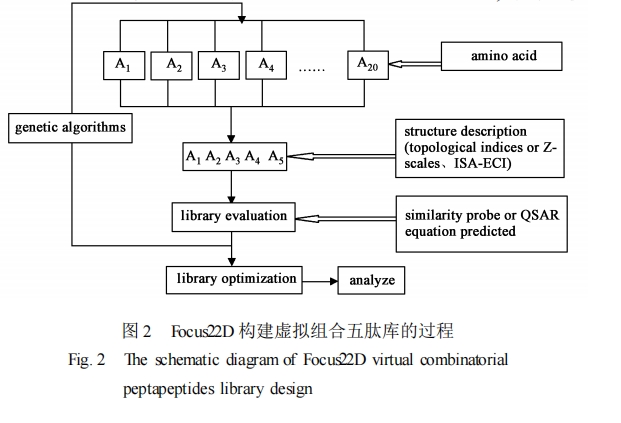
利用虚拟组合多肽库的最终目的是为了发现先导化合物 ,而且要求合成出的这些化合物能够为先导化合物的发现和优化提供尽可能多的构效关系信息。也就是说在保证多肽库体积的前提下 ,根据实验目的设计出满足一定分子多样性的组合多肽库。可见 ,随着计算机辅助分子设计和基于结构的药物设计方法在虚拟组合多肽库中的应用 ,虚拟组合多肽库的质量和筛选效率会得到更大地提高 ,而且虚拟组合多肽库在多肽药物的分子设计中 ,将会提供更多的理论依据和指导思想。
三、结 语
可以预见 ,随着计算机辅助分子设计的发展 ,多肽的定量构效关系、模拟肽学和虚拟组合多肽库会更多地用于指导先导化合物的研究和开发 ,而且多肽类药物分子设计方法会更加完善和丰富 ,相信在不久的将来此研究领域会有更大的突破。
免责声明:本文为行业交流学习,版权归原作者及原杂志所有,如有侵权,可联系删除。文章标注有作者及文章出处,如需阅读原文及参考文献,可阅读原杂志。

电话:0551-65177703 邮箱:pb@peptidesbank.com 地址:安徽省合肥市四川路868号云谷创新园A6栋3层
合肥肽库生物(Taikubio)只为有资质的科研机构、医药企业基于科学研究或药证申报的用途提供医药研发服务, 不为任何个人或者非科研性质的、非用于药证申报使用等其他用途提供服务。