
’Σ“ΣΘΚΒΑΑΉΕύκΡ“©ΈοΨΏ”–ΝΦΚΟΒΡΧΊ“λ–‘ΚΆ…ζΈοœύ»ί–‘Θ§÷ΈΝΤ–ßΙϊΫœΚΟΓΘ”…”ΎΩΎΖΰΗχ“©ΆΨΨΕΒΡΜΦ’ΏΥ≥”Π–‘ΚΆΑ≤»Ϊ–‘ΗϋΦ―Θ§ΒΑΑΉΕύκΡ“©ΈοΒΡΩΎΖΰΒίΥΆ“―≥…ΈΣΒ±«Α≤ΡΝœ―ß”κ“©ΦΝ―ßΝλ”ρΒΡ―–ΨΩ»»Βψ÷°“ΜΓΘ»ΜΕχΘ§ΩΎΖΰ…ζΈο¥σΖ÷Ή”“©Έο‘ΎΈΗ≥ΠΒάΒΡΈ»Ε®–‘ΦΑΈϋ ’≥ΧΕ»»¥Ζ«≥Θ ήœόΓΘ÷§÷ Ρ…ΟΉ‘ΊΧεΡήΆ®ΙΐΜ·―ß–ό ΈΓΔ ηΥ°άκΉ”≈δΕ‘Β»Εύ÷÷ΖΫ Ϋ”––ßΑϋ‘Ί«Ή÷§Μρ«ΉΥ°–‘ΒΑΑΉΕύκΡ“©ΈοΘ§Ά§ ±Ψ≠±μΟφ–ό ΈΚσΒΡ÷§÷ Ρ…ΟΉ‘ΊΧεΡήΩΥΖΰΩΎΖΰΈϋ ’ΒΡ÷ΎΕύ…ζάμΤΝ’œΘ§ΦΧΕχ¥ΌΫχ“©Έο‘ΎΜλΚœΫΚ χ÷–»ήΫβΘ§‘ω«ΩΝήΑΆ…ψ»ΓΘ§“ρ¥ΥΨΏ”–ΝΦΚΟ…ζΈοœύ»ί–‘ΚΆΧεΡΎΈ»Ε®–‘ΓΘ±Ψ―–ΨΩΉέ ωΝΥ÷§÷ Ρ…ΟΉ‘ΊΧεΕ‘ΒΑΑΉΕύκΡ“©ΈοΒΡΑϋ‘ΊΖΫ ΫΦΑΤδΩΥΖΰ…ζάμΤΝ’œΒΡœύ”ΠΜζ÷ΤΘ§Ϋι…ήΝΥΤδΧαΗΏΒΑΑΉΕύκΡ“©ΈοΩΎΖΰ…ζΈοάϊ”ΟΕ»ΒΡ÷Ί“ΣΧΊ–‘ΚΆΉν–¬―–ΨΩΫχ’ΙΘ§≤Δ’ΙΆϊΝΥΤδ«±‘Ύ”Π”ΟΦΑΖΔ’Ι«ΑΨΑΓΘ
ΒΑΑΉΕύκΡ“©Έο( protein and peptide drugsΘ§PPDs) ΨΏ”–…ζΈοΜν–‘ΗΏΓΔΧΊ“λ–‘«ΩΓΔ»ήΫβ–‘«ΩΓΔΕΨ–‘ΒΆΒ»”≈ΒψΘ§Ά®ΙΐΒςΫΎ…ζάμΜρ≤ΓάμΙΐ≥Χά¥÷ΈΝΤΜρ‘ΛΖάΦ≤≤ΓΘ§ΥφΉ≈…ζΈοΦΦ θΒΡ―ΗΥΌΖΔ’ΙΘ§‘ΎΝΌ¥≤Φ≤≤Γ÷ΈΝΤ…œ‘Ϋά¥‘ΫΕύΒΊ±ΜΩΣΖΔ”Π”ΟΓΘΨΓΙήΖ«ΈΗ≥ΠΒάΗχ“© «PPDs Ήν≥Θ”ΟΒΡΗχ“©ΆΨΨΕΘ§ΒΪΜΦ’ΏΥ≥”Π–‘ΆυΆυΫœ≤νΓΘ”κΨ≤¬ωΗχ“©œύ±»Θ§ΩΎΖΰΗχ“©ΆΨΨΕ‘ΎΜΦ’ΏΥ≥”Π–‘ΓΔΑ≤»Ϊ–‘ΓΔ≥ΛΤΎΦΝΝΩΚΆ÷Τ‘λ≥…±ΨΖΫΟφΨΏ”–”≈ Τ [1]ΓΘ»ΜΕχΘ§”κ–ΓΖ÷Ή”“©Έοœύ±»Θ§PPDs ΒΡΙψΖΚ”Π”Ο ήΒΫΝΥΈ»Ε®–‘ΚΆΈϋ ’≥ΧΕ»ΒΡœό÷ΤΓΘ
–¬–ΆΩΎΖΰ¥ΌΈϋ ’ΦΦ θΒΡΩΣΖΔ“―≥…ΈΣΧαΗΏ PPDs…ζΈοάϊ”ΟΕ»ΒΡ“Μ÷÷ΖΫ ΫΘ§ΡΩ«Α“―ΆΤ≥ωΒΡΩΎΖΰΕύκΡΚΆΒΑΑΉ÷ ÷ΤΦΝΒΡ ΐΝΩ’ΐ‘Ύ≤ΜΕœ‘ωΦ”ΓΘ2001 ÷Ν 2006 ΡξΟάΙζ Emisphere ΙΪΥΨΫχ––ΝΥ ΉΗωΩΎΖΰ“»ΒΚΥΊ÷ΤΦΝΔώΤΎΚΆΔρΤΎΝΌ¥≤ ‘―ιΘ§―–ΨΩœ‘ ΨΩΎΖΰ÷ΤΦΝ”κΑ≤ΈΩΦΝΟΜ”–œ‘÷χ≤ν“λΓΘ2014 ΡξΘ§”…“‘…ΪΝ– Oramed ΙΪΥΨΩΣΖΔΒΡ ORMD-0801 ±ΜΟάΙζ FDA ≈ζΉΦΫχ––ΔσΤΎΝΌ¥≤ ‘―ιΓΘEmisphere ΙΪΥΨΩΣΖΔΝΥ“Μ÷÷ΩΎΖΰΈϋ ’¥ΌΫχΦΝ N-[8-(2- τ«±ΫΦΉθΘΑ±Μυ ) ] –ΝΥαΡΤ (SNAC)Θ§œ»±Μ”Ο”ΎΗΡ…Τ“»ΒΚΥΊΒΡΩΎΖΰΈϋ ’Θ§Ήν÷’±Μ”Ο”ΎΗΡ…ΤΥς¬μ¬≥κΡΒΡΩΎΖΰΈϋ ’ [2]ΓΘ2019 ΡξΘ§”…ΒΛ¬σ≈ΒΚΆ≈ΒΒ¬ΙΪΥΨΩΣΖΔΒΡ»Ϊ«ρ ΉΗωΩΎΖΰ“»ΗΏ―ΣΧ«ΥΊ―υκΡ -1(GLP-1) ήΧεΦΛΕ·“©Υς¬μ¬≥κΡ ( …ΧΤΖΟϊ Rybelsus) ±ΜΟάΙζ FDA ≈ζΉΦ”Ο”Ύ÷ΈΝΤ 2 –ΆΧ«Ρρ≤ΓΓΘΥφΚσ‘Ύ 2020 ΡξΆ®Ιΐ“ΐ»κΥ≤ ±…χΆΗ–Ά‘ω«ΩΦΝ (transient permeability enhancerΘ§TPE) ΦΦ θ÷Τ±ΗΝΥΑ¬«ζκΡΩΎΖΰ≥Π»ήΫΚΡ“( …ΧΤΖΟϊ Mycapssa)Θ§“≤ΜώΒΟΝΥΟάΙζ FDA ΒΡ≈ζΉΦ [3]ΓΘΡΩ«Α’κΕ‘ PPDs ΒΡΩΎΖΰΗχ“©œΒΆ≥Θ§÷ΎΕύ―ß’ΏΜΙΙΙΫ®ΝΥΥ°ΡΐΫΚΓΔΈΔ«ρΓΔΡ…ΟΉΝΘΓΔ÷§÷ Ρ…ΟΉΝΘΒ»Εύ÷÷ΒίΥΆ≤Ώ¬‘Θ§“‘ΧαΗΏ“©ΈοΒΡΩΎΖΰ…ζΈοάϊ”ΟΕ»ΓΘ‘ΎΙΐ»ΞΦΗ °Ρξ÷–Θ§Ρ…ΟΉ‘ΊΧεΦΦ θ“ρΩ…‘ωΦ”…ζΈοΡΛΆΗΙΐ–‘ΓΔΗΡ±δ“©ΈοΧεΡΎΖ÷≤ΦΓΔΒςΫΎ Ά“©ΥΌΕ»Β»ΙΠΡήΕχ‘Ύ“©ΤΖΓΔΜ·Ή±ΤΖΒ»Νλ”ρ÷–’Ιœ÷≥ωΨό¥σ«±ΝΠΘ§ΩΎΖΰΒίΥΆ PPDs ΒΡΡ…ΟΉ‘ΊΧε÷πΫΞ≥…ΈΣ―–ΨΩ»»ΒψΓΘΫϋΤΎ”–―–ΨΩ…ηΦΤ Ι”ΟΡΆΥαΫπ τ - ”–ΜζΙ«ΦήΡ…ΟΉΝΘά¥Αϋ‘ΊΉψΙΜΦΝΝΩΒΡ“»ΒΚΥΊΘ§≤Δ‘Ύ±μΟφ–ό ΈΑ–ΒΑΑΉΘ§“‘ Βœ÷ΗΏ–ßΒΡ“»ΒΚΥΊΩΎΖΰΗχ“©Θ§‘≠άμ «Ά®Ιΐ ήΧεΫιΒΦΒΡΩγœΗΑϊΆΨΨΕΘ§ ΙΗΟΡ…ΟΉΝΘΡή Βœ÷ΗΏ–ßΒΡ≥Π…œΤΛΉΣ‘ΥΘ§≤Δ‘Ύ…ζάμΧθΦΰœ¬ΩΊ÷Τ“»ΒΚΥΊΒΡ ΆΖ≈Θ§ΨΏ”–œ‘÷χΒΡΫΒΧ«Ής”Ο [4]ΓΘ
Ε‘”Ύ PPDs ΒΡΩΎΖΰΒίΥΆΘ§άμœκΒΡΡ…ΟΉ‘ΊΧε”ΠΨΏ±Η”≈ΝΦΒΡΈ»Ε®–‘ΚΆ…ζΈοœύ»ί–‘ΓΔΫœΗΏΒΡ‘Ί“©ΝΩΒ»ΧΊ–‘ΓΘ20 άΦΆ 90 Ρξ¥ζ÷§÷ Ρ…ΟΉ‘ΊΧε ( lipid nanocarriersΘ§LN) ΖΔ’Ι≥… λΘ§Υϋ“‘…ζΈοœύ»ίΒΡ÷§÷ ≤ΡΝœ ( »γΗ ”Ά»ΐθΞΚΆ÷§ΖΨΥα ) ΈΣ‘ΊΧεΘ§ΫΪ“©Έο»ήΫβΜρΑϋΙϋ‘Ύ÷§÷ ΚΥ–ΡΜρΈϋΗΫ”ΎΡ…ΟΉΝΘ±μΟφΘ§Αϋά®ΙΧΧε÷§÷ Ρ…ΟΉΝΘ (solid lipid nanoparticlesΘ§SLNs)ΓΔΡ…ΟΉΫαΙΙ÷§÷ ‘ΊΧε (nanostructured lipid carriersΘ§NLCs)Β»Θ§ΨΏ”–Φθ…ΌΈΗ≥ΠΒάΟΗΫβΓΔ¥ΌΫχ–Γ≥Π…œΤΛΈϋ ’ΓΔ…ζΈοœύ»ί–‘ΚΟΒ»”≈ ΤΘ§ Κœ…ζΈο¥σΖ÷Ή”άύ“©ΈοΒΡΩΎΖΰΗχ“©Θ§≤ΔΨΏ”–ΙΛ“ΒΜ·…ζ≤ζΒΡΩ…Ρή [5ΓΣ6]ΓΘ±Ψ―–ΨΩΉήΫαΝΥ PPDs ΈΗ≥ΠΒάΈϋ ’ΒΡ’œΑ≠Μζ÷ΤΦΑΩΎΖΰΒίΥΆ≤Ώ¬‘ΓΔ÷§÷ Ρ…ΟΉΒίΥΆ‘ΊΧε¥ΌΫχ PPDs ΩΎΖΰΈϋ ’Μζ÷ΤΦΑΤδ”Ο”ΎΩΎΖΰΒίΥΆ PPDs ΒΡ”Π”ΟΫχ’ΙΘ§≤ΔΫχ“Μ≤ΫΧΫΧ÷ΤδΧτ’Ϋ”κΜζ”ωΓΘ
1 PPDs ΩΎΖΰΗχ“©œ÷Ή¥
1.1 PPDs ΈΗ≥ΠΒάΈϋ ’’œΑ≠Μζ÷Τ
‘ΎΈΗ≥ΠΒά÷–Θ§PPDs “ΜΖΫΟφ“Ή ήΈΗΥαΫΒΫβΤΤΜΒΘ§ΜρΗΡ±δΫβάκΉ¥Χ§Θ§ΒΦ÷¬ΫαΙΙΜρΙΠΡήΗΡ±δΘ§Νμ“ΜΖΫΟφΗς÷÷œϊΜ·ΟΗ“≤ΜαΫΒΫβ PPDs[7]ΓΘΆ§ ±Θ§≤ΩΖ÷ PPDs ”…”Ύ«ΉΥ°–‘Ϋœ«Ω Θ§ΈόΖ®”––ߥ©ΆΗ–Γ≥ΠπΛ“Κ≤ψΚΆœΗΑϊΦδΫτΟήΝ§Ϋ”ΒΡ…œΤΛ≤ψ ( œΒ PPDs Έϋ ’Ιΐ≥ΧΒΡ 2 ΒάΈοάμΤΝ’œΘ§«Α’ΏΉηΑ≠“©ΈοœρΈϋ ’ΡΛ±μΟφά©…ΔΘ§Κσ’Ώ–Έ≥…ΝΥ“÷÷Τ“©Έο…χΆΗΒΡΟήΖβ±ΎάίΘ§ΒΦ÷¬ΡΛΆ®ΆΗ–‘Ϋœ≤ν [8ΓΣ9])Θ§ΒΦ÷¬ PPDs ΚήΡ―±ΜΈϋ ’ [10]
1.2 PPDs ΩΎΖΰΒίΥΆΒΡ“ΜΑψ≤Ώ¬‘
1.2.1 Μ·―ß–ό Έ
PPDs ΩΎΖΰ…ζΈοάϊ”ΟΕ»ΆυΆυ ήΒΫ“©ΈοΉ‘…μ«ΉΥ°–‘ΓΔœύΕ‘Ζ÷Ή”÷ ΝΩΦΑΕ‘ΟΗΚΆ pH ΟτΗ–Β»άμΜ·–‘÷ ΒΡ”ΑœλΘ§“ρ¥Υ≥Θ≤…”ΟΜ·―ß–ό Έ≤Ώ¬‘ά¥ΗΡ±δ PPDs ΒΡœύ”Π–‘÷ [11ΓΣ12]ΓΘ Ήœ»ΩΦ¬«Ά®ΙΐΧμΦ”Ζ«ΦΪ–‘ΜυΆ≈Μρ»Ξ≥ΐΦΪ–‘ΜυΆ≈ά¥‘ωΦ”“©ΈοΒΡ«Ή÷§–‘Θ§“‘άϊ”Ύ“©Έο‘Ύ≥ΠπΛΡΛ…œΒΡά©…ΔΘ§ΒΪ PPDs ΒΡΥ°»ή–‘ΫΒΒΆΜαΒΦ÷¬”κ ήΧεΒΡ«ΉΚΆΝΠΫΒΒΆΘ§Ι ΜΙ–ηΗ®“‘ΨέΚœΈο≤ΡΝœΫχ––”≈Μ·ΓΘΤδ¥ΈΘ§”…”Ύ≥ΠΒάœΗΑϊΡΛ…œΒΡΧ«ΒΑΑΉ¥χΗΚΒγΚ…Θ§―τάκΉ”“©ΈοΕ‘≥ΠπΛΡΛΒΡΆ®ΆΗ–‘Ηϋ«Ω [13ΓΣ14]ΓΘΒΪΕύκΡ―τάκΉ”Μ·Ω…ΡήΜα≤ζ…ζΟβ“Ώ‘≠–‘ΦΑ«±‘ΎΕΨ–‘ΓΘΉνΚσΘ§Ψέ““Εΰ¥Φ (PEG) Μ·Ω…Ά®ΙΐΩ’ΦδΈΜΉηΕχΉηΒ≤ PPDs ”κΒΑΑΉΥ°ΫβΟΗΒΡΫαΚœΘ§―”≥ΛΤδΑκΥΞΤΎ [15]Θ§“≤ «‘ωΦ”“©ΈοΈϋ ’ΒΡ“ΜΗω”––ßΆΨΨΕΘ§ΒΪœύΕ‘Ζ÷Ή”÷ ΝΩΒΡ‘ωΦ”“≤ΜαΒΦ÷¬Ω≈ΝΘ‘ω¥σΓΔπΛΕ»‘ωΦ”ΦΑœΗΑϊ«ΉΚΆΝΠΫΒΒΆΒ»Έ ΧβΓΘΡΩ«ΑΘ§÷§ΜυΩ≈ΝΘΓΔΕύΧ«ΜυΩ≈ΝΘΒ»ΕύΙΠΡήΩ≈ΝΘœΒΆ≥ΡήΆ®ΙΐΕύ÷ΊΜ·―ß–ό ΈΕχ Βœ÷÷ΈΝΤ–‘ PPDs ΒΡ”––ßΩΎΖΰΒίΥΆ [16]ΓΘ
1.2.2 Φ”»κ”––ßΗΡ–‘ΦΝ
Έϋ ’¥ΌΫχΦΝΆ®Ιΐ¥ΌΫχ“©ΈοΕ‘≥ΠΒάœΗΑϊΒΡ…χΆΗΕχΗΡ…Τ“©ΈοΈϋ ’Θ§“ρ¥Υ‘ΎΧαΗΏ…ζΈοάϊ”ΟΕ»ΒΡΆ§ ±“≤Ω…ΡήΜαΤΤΜΒΡΛΆξ’ϊ–‘ΓΔ≤ζ…ζ»Ϊ…μΕΨ–‘ [17]ΓΘPPDs ÷ΤΤΖΆ®≥ΘΜαΑϋΙϋ≥Π»ή≤ψΘ§“‘Ζά÷Ι‘ΎΥα–‘ΜΖΨ≥÷–ΫΒΫβΓΘ―–ΨΩ’Ώ“―÷ΛΟςΘ§ΝΣΚœΖΰ”ΟηέιΎΥαΩ…ΫΒΒΆ≥ΠΒά“»ΒΑΑΉΟΗΒΡΜν–‘Θ§≤Δ¥ΌΫχΫΒΗΤΥΊΒΡΩΎΖΰΈϋ ’ [18]ΓΘΆ®Ιΐ Ι”ΟΟΗ“÷÷ΤΦΝ÷±Ϋ”“÷÷ΤΒΑΑΉΥ°ΫβΟΗΒΡΜν–‘ «»ΤΙΐ≥ΠΒάΟΗΫΒΫβΒΡΝμ“Μ÷÷ΖΫ ΫΓΘΉν–¬ΝΌ¥≤―–ΨΩ÷–ΒΡΟΗ“÷÷ΤΦΝΑΗάΐ «÷ΊΉι»Υ“»ΒΚΥΊ≥Π»ήΫΚΡ“ (ORMD-0801)Θ§Υϋ”…¥σΕΙ“»ΒΑΑΉΟΗ“÷÷ΤΦΝΚΆ“Μ÷÷«ε≥ΐΗΤΒΡρϋΚœΦΝΉι≥…Θ§ ‘―ιœ‘ Ψ’β÷÷÷ΈΝΤΖΫΑΗΡήœ‘÷χΫΒΒΆ―ΣΧ«Θ§”––ßΧαΗΏΩΎΖΰ…ζΈοάϊ”ΟΕ» [19]ΓΘΨ≠πΛ“Κ¥©ΆΗΦΝ–ό ΈΒΡΡ…ΟΉΝΘ‘Ύ…œΤΛπΛ“Κ≤ψ÷–Ω…±μœ÷≥ωΉ‘”…ΒΡ≤Φά ‘ΥΕ·Θ§»γ Ι”ΟN-(2- τ«±ϊΜυ ) ΦΉΜυ±ϊœ©θΘΑΖΨέΚœΈο ( pHPMA) ΉςΈΣΩ…ΫβάκΒΡπΛ“ΚΕη–‘ ‘ΦΝΩ…¥ΌΫχΡ…ΟΉΝΘΒΡπΛ“Κ…χΆΗΘ§‘ΎœΗΑϊ¥©ΆΗκΡ (CPP) ΒΡΫιΒΦœ¬ΧαΗΏ…œΤΛΈϋ ’ΡήΝΠ[20ΓΣ21]ΓΘ”––ßΒΡΗΡ–‘ΦΝ¥σ÷¬Αϋά®±μ1÷–ΒΡ5άύΈο÷ ΓΘ

2 LN Βœ÷Αϋ‘Ί«ΉΥ°–‘ PPDs ΒΡΕύ÷ΊΜζ÷Τ
2.1 ¥ΪΆ≥Ι≤Φέ–ό Έ
ΈΣΝΥΫΪ«ΉΥ°–‘ PPDs ’ϊΚœΒΫ LN ÷–Θ§±Ί–κ…ηΖ®ΧαΗΏΤδ«Ή÷§–‘Θ§Εχ¥ΪΆ≥ΒΡΜ·―ß–ό ΈΖΫΖ®Ω…ΧαΙ©“Μ–©ΥΦ¬ΖΓΘ Ήœ»Θ§¥σΕύ ΐΕύκΡΕΦ÷Ν…ΌΚ§”–“ΜΗω≤°Α±ΜυΓΔτ«ΜυΜρτ»ΜυΙΌΡήΆ≈Θ§Ω…Ά®ΙΐθΞΦϋΜρθΘΑΖΦϋ”κ÷§ΖΨΥαΙ≤ΦέΫαΚœΓΘ¥ΥΆβΘ§ΕΰΝρΦϋ“≤Ω…±Μ”ΟΉςΝ§Ϋ”±έ [22]ΓΘΕύκΡΜΖΜ·“≤ΡήΗΡ±δΕύκΡΒΡάμΜ·–‘÷ Θ§ ΙΙΙœσΗ’–‘±δ«ΩΕχ–Έ≥…Ζ÷Ή”ΡΎ«βΦϋΘ§«Ή÷§–‘Υφ÷°‘ωΦ”Θ§Ά§ ±«Ή÷§–‘ΒΡΖ«¥χΒγΜΖκΡΩ…Ά®ΙΐΩγœΗΑϊΆΨΨΕ±ΜΕ·ά©…Δ¥©ΆΗœΗΑϊΡΛ [23]ΓΘ»ΜΕχΘ§’β–©ΖΫΖ®–η“ΣΗ¥‘”ΒΡΚœ≥…ΚΆ¥ΩΜ·≤Ϋ÷ηΘ§Ά§ ±Μ·―ß–ό Έ≤ζ…ζΒΡ–¬Μν–‘“©Έο≥…Ζ÷±Ί–κΆ®ΙΐΦύΙήΜζΙΙΒΡ…σ≈ζΘ§œύΕ‘ά¥ΥΒΖ«Ι≤ΦέΫαΚœΒΡΕύκΡΩ…Ρή «Ϋœ”≈―Γ‘ώ [24]ΓΘ
2.2 ηΥ°άκΉ”≈δΕ‘ (hydrophobic ion pairingΘ§HIP)
Ζ«Ι≤ΦέΫαΚœΉς”ΟΑϋά®«βΦϋΓΔ ηΥ°Ής”ΟΚΆάκΉ”œύΜΞΉς”ΟΒ»Θ§Τδ÷–Ϋœ”––ßΒΡΖΫΖ® «άκΉ”œύΜΞΉς”ΟΘ§Φ¥Ά®Ιΐ“θάκΉ”ΓΔΝΫ–‘Μρ―τάκΉ”±μΟφΜν–‘ΦΝ÷–ΚΆΕύκΡΒΡΨΜΒγΚ…ά¥‘ωΦ”ΕύκΡ«Ή÷§–‘ [22]ΓΘ
HIP ΉςΈΣ“Μ÷÷ΫΪ¥χΒγΒΡ«ΉΥ°Ζ÷Ή””κ¥χœύΖ¥ΒγΚ…ΒΡΖ¥άκΉ”Ά®ΙΐάκΉ”œύΜΞΉς”ΟΫαΚœΉΣ±δΈΣ ηΥ°¬γΚœΈοΒΡ≤Ώ¬‘ΕχΒΟΒΫΙψΖΚΙΊΉΔΓΘΖ¥άκΉ”÷Ν…ΌΑϋΚ§“ΜΗω ηΥ°ΫαΙΙ”ρΘ§Ής”ΟΙΐ≥Χ÷–«ΉΥ°Ζ÷Ή”ΒΡΒγΚ…±Μ ηΥ°ΫαΙΙ”ρΤΝ±ΈΘ§Φθ…ΌΝΥ‘ΎΦΪ–‘»ήΦΝ÷–ΒΡ»ήΫβΕ» ΘΜΕχΕύκΡ÷Ν…ΌΚ§”–“Μ÷÷Ω…ΒγάκΒΡΑ±ΜυΥαΘ§Ά®ΙΐΒς÷ΝΒ»ΒγΒψ“‘…œΒΡ pH ÷ΒΘ§ ΙΕύκΡ≤ζ…ζΗΚΒγΚ…Θ§Ζ¥÷°‘ρ≤ζ…ζ’ΐΒγΚ…ΓΘ“ρ¥ΥΘ§HIP Ω…”––ßΧαΗΏ«ΉΥ°–‘ΕύκΡΒΡ«Ή÷§–‘≤ΔΫΪΤδΖβΉΑΒΫΡ…ΟΉ‘ΊΧε÷– [25]ΓΘ
“ΜœΒΝ–―–ΨΩ±μΟςΘ§άϊ”Ο HIP ‘≠άμΩ…‘ωΦ”ΕύκΡΒΡ«Ή÷§–‘Θ§ ΙΤδ»ήΫβ‘Ύ”Άœύ÷–ΓΘΜυ”Ύ“θάκΉ”±μΟφΜν–‘ΦΝΚΆ’ΐΒγΚ…ΒΑΑΉ÷ ΒΡœύΜΞΉς”ΟΘ§―–ΨΩ’ΏΆ®Ιΐ HIP ΫΪ“»ΒΚΥΊΑϋΙϋ‘Ύ”≤÷§ΥαΙΧΧε÷§÷ Ρ…ΟΉΝΘ÷– [26]ΓΘΈΣΜώΒΟΫœΗΏΒΡ”––ß‘ΊΚ…Θ§”–―ß’Ώ―–ΨΩΝΥΝΝ±ϊ»πΝ÷ (LEU)ΓΔ“»ΒΚΥΊ ( INS) ΚΆ»ΞΑ±Φ”―ΙΥΊ (DES) Β»―τάκΉ”ΕύκΡ”κ“θάκΉ”±μΟφΜν–‘ΦΝΕΰ °ΕΰΆιΜ«ΥαΡΤΓΔ °ΕΰΆιΜυΝρΥαΡΤ (SDS) ΚΆ”ΆΥαΡΤΒΡ HIP ¬γΚœΈο‘ΎΗς÷÷»ήΦΝ÷–ΒΡ»ήΫβΕ»Θ§÷ΛΟςΕύκΡ“©ΈοΒΡ”––ß‘ΊΚ…Ω…¥οΒΫ 10ΘΞ“‘…œ [27]ΓΘNAZIR Β»Ϋχ“Μ≤ΫΩΦ¬«ΝΥ Lipinski ΈεΖ®‘ρΦΑ«βΦϋΙ©ΧεΚΆ ήΧε―«ΫαΙΙΕ‘ΡΛΆ®ΆΗ–‘ΒΡ”ΑœλΘ§»œΈΣΧαΗΏΕύκΡ«Ή÷§–‘ΒΡΙΊΦϋ“ρΥΊ «ΫΒΒΆΤδ«βΦϋ«±ΝΠΘ§“ρ¥ΥΧα≥ωΝΥ ηΥ°«βΦϋ≈δΕ‘ (hydrophobic H-bond pairingΘ§HHP) Ζ®Θ§Φ¥«ΉΥ°–‘¥σΖ÷Ή”“©ΈοΆ®Ιΐ«βΦϋ”κΖ«άκΉ”±μΟφΜν–‘ΦΝ ( »γ“‘’αΧ«ΈΣ«ΉΥ°–‘ΆΖΜυΓΔ÷§ΖΨΥαΈΣ«Ή÷§–‘Έ≤Μυ ) –Έ≥…«Ή÷§–‘Η¥ΚœΈοΘ§¥”ΕχΗΡ…ΤΡΛΆ®ΆΗ–‘Θ§≤Δ÷ΛΟς HHP Ά®Ιΐ“ΐ»κ«Ή÷§–‘―«ΫαΙΙΚΆœϊ≥ΐ«βΦϋΙ© ήΧε―«ΫαΙΙΕ‘ΗΡ…Τ LEU ΒΡ«Ή÷§–‘ΚΆΡΛ…χΆΗ–‘”–Ϋœ¥σΉς”Ο [24]ΓΘ
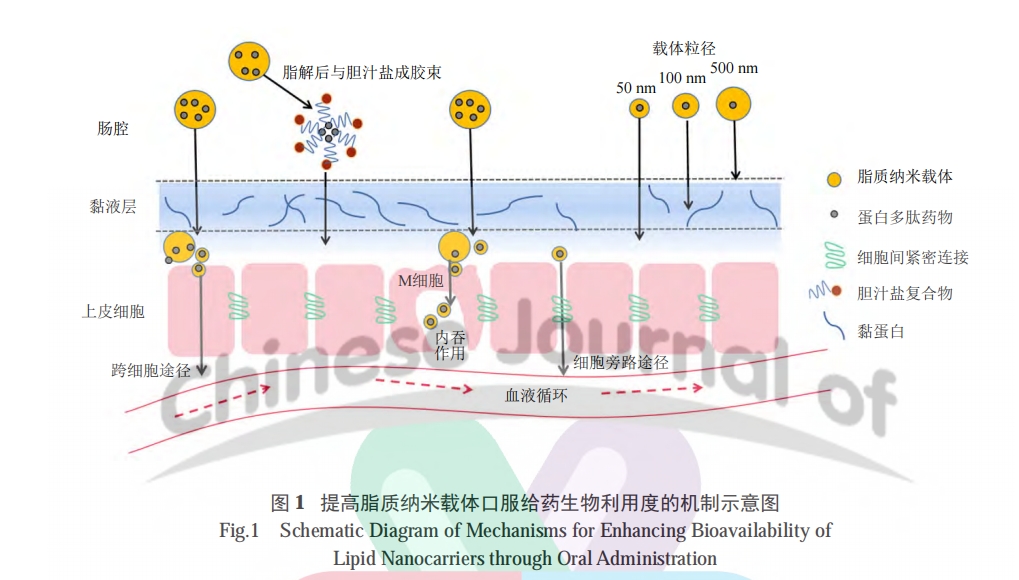
3 LN ΧαΗΏ PPDs ΩΎΖΰ…ζΈοάϊ”ΟΕ»ΒΡΜζ÷Τ
3.1 LN ΩΥΖΰ…ζάμ–‘ΤΝ’œ
ΩΎΖΰ PPDs ΟφΝΌΒΡ÷ς“Σ’œΑ≠ «ΈΗΥαΚΆΟΗΘ§ΜαΒΦ÷¬ PPDs ΧΊΕ®ΫαΙΙΚΆΙΠΡήΒΡ…Ξ ß [28]ΓΘΨ≠Ιΐ Β±Αϋ“¬ΒΡ LN “―±Μ÷ΛΟςΩ…±ήΟβΈΗΥαΦΑΒΑΑΉΟΗΫΒΫβΘ§Ά§ ±HIP ΒΡ–Έ≥…¥σΖυ‘ωΦ”ΝΥ PPDs ΒΡ«Ή÷§–‘Θ§ ΙΤδΩ…»ήΫβ‘ΎΡ…ΟΉ‘ΊΧεΒΡ«Ή÷§œύ÷–ΓΘ«“ HIP ≤Μ ήΨΚ’υ–‘Ζ¥άκΉ”ΒΡ”ΑœλΘ§ΕχΟΗ”…”Ύ«ΉΥ°ΧΊ–‘≤ΜΡήΫχ»κ«Ή÷§œύΘ§“ρ¥Υ÷ΈΝΤ–‘ PPDs ‘Ύ LN ÷–Ω… ήΒΫ±ΘΜΛΓΔ≤Μ±ΜΫΒΫβ [15]ΓΘ
“ΜΒ©Ϋχ»κ–Γ≥ΠΘ§¥σΕύ ΐ LN Μα±Μ≥Π“Κ÷–ΒΡ÷§ΖΨΟΗ―ΗΥΌΫΒΫβΓΘARNOLD Β»Ζ÷ΈωΝΥ≤ΜΆ§Η®Νœ ( ¥”»ΐΗ θΞΓΔΥΪΗ θΞΚΆΒΞΗ θΞΒΡΜλΚœΈοΒΫΖ«άκΉ” PEG Μ·ΚΆΨέΗ ”Ά±μΟφΜν–‘ΦΝ ) Ε‘÷§ΖΨΟΗΒΡΫΒΫβ––ΈΣ [29]ΓΘ“Μ–©ΨΏ”–“Ή±Μ÷§ΖΨΟΗΓΔΒΑΑΉΟΗ»Ξ≥ΐθΞΫαΙΙΒΡΗ®Νœ”Π±ήΟβ”Ο”Ύ÷Τ±Η LNΓΘLEONAVICIUTE Β»÷ΛΟςθΞάύΒΡ”ΟΝΩΚΆ÷÷άύΩ…ΉΦ»ΖΒςΫΎΉ‘»ιΜ· Ά“©œΒΆ≥«Ή÷§œύΒΡΟΗ¥ΌΫΒΫβΘ§LN ΒΡΫΒΫβ«ζœΏΩ…¥”ΩλΥΌΫΒΫβ (<1 h) Βς’ϊΒΫΆξ»Ϊ≤ΜΫΒΫβ [30]ΓΘLN ΒΡ±μΟφ–ό ΈΕ‘Τδ…ζΈοΫΒΫβ–‘“≤”–”ΑœλΘ§ΫΪΚ§”–÷–Ν¥Η ”Ά»ΐθΞΒΡ÷§ΜυΡ…ΟΉ‘ΊΧε”ΟPEG ΑϋΗ≤Θ§Ω…ΫΒΒΆ÷§ΖΨΟΗΒΡœϊΜ·Θ§’β÷÷ΤΝ±Έ–ß”ΠΥφΉ≈ PEG Ν¥≥ΛΕ»ΒΡ‘ωΦ”Εχ‘ωΦ”ΓΘ
”–―–ΨΩΤάΙάΝΥ”Ο”Ύ NLCs ΒΡ±μΟφΜν–‘ΦΝΕ‘PPDs ΧαΙ©ΟΗ±ΘΜΛΒΡ”Αœλ [31]ΓΘ Ήœ»ΫΪ“»ΒΚΥΊΉςΈΣΡΘ–ΆκΡ”κ SDS Ά®ΙΐάκΉ”Φϋ–Έ≥…Η¥ΚœΧε ( INS-SDS)“‘ΧαΗΏ«Ή÷§–‘ ( “»ΒΚΥΊΒΡ logP ¥” ®C1.8 …ΐ÷Ν 2.1)ΓΘ»ΜΚσΆ®Ιΐ»ήΦΝά©…ΔΖ®÷Τ±ΗΝΥ 3 ÷÷ NLCs[ Ζ÷±πΚ§”–PEG θΞΓΔPEG Ο―ΚΆΨέΗ ”ΆθΞ (PG θΞ ) ]Θ§‘ΌΫχ––ΧεΆβ÷§ΫβΚΆΡΘΡβΈΗ≥Π“Κ÷–ΒΡΒΑΑΉ÷ Υ°Ϋβ―–ΨΩΓΘΫαΙϊ±μΟςΘ§Κ§ PEG Ο―ΒΡ NLCs Ε‘“»ΒΚΥΊΗ¥ΚœΧεΒΡ±ΘΜΛ–ßΙϊΉνΚΟΘ§’β «“ρΈΣ÷ΤΤΖ÷–»±ΖΠθΞΜυΫαΙΙΘ§≤Μ“Ή ή“»ΟΗΓΔ÷§ΖΨœϊΜ·ΟΗΒΡ”ΑœλΓΘΒΑΑΉΫΒΫβ―–ΨΩ±μΟςΘ§INS-SDSΗ¥ΚœΖ÷Ή”Ρή±Μ“»ΟΗΆξ»ΪΫΒΫβΘ§Εχ±Μ NLCs ΑϋΖβΚσ”κ“»ΟΗΙ≤Ζθ”ΐ 4 hΘ§»‘”– 30ΘΞΓΪ 50ΘΞΒΡΕύκΡΈ¥±ΜΫΒΫβΓΘNLCs ±μΟφΚ§”–Ε‘ΫΒΫβΟΗ≤ΜΟτΗ–ΒΡ―«ΫαΙΙΘ§Ω…ΈΣ PPDs ΧαΙ©ΗϋΚΟΒΡΈΗ≥ΠΒάΒΑΑΉΟΗ±ΘΜΛΓΘ
–Γ≥Π «ΕύκΡΚΆΒΑΑΉ÷ œϊΜ·ΒΡ÷ς“Σ≤ΩΈΜΘ§“ρ¥Υ≥ΠπΛΡΛΦΑ…œΤΛ « PPDs ΩΎΖΰΒίΥΆΒΡ’œΑ≠ [28]ΓΘLN ΒΡ«ΉΥ°–‘ΚΆ±μΟφΒγΚ… «”Αœλ LN …χΆΗ–Γ≥Π…œΤΛœΗΑϊΒΡΙΊΦϋ“ρΥΊΓΘœ»«Α―–ΨΩ±μΟςΘ§¥χ’ΐΒγΚ…ΒΡ LN( »γΩ«ΨέΧ«–ό ΈΒΡ SLNs) Ε‘πΛ“Κ≤ψΨΏ”–ΗϋΗΏΒΡ«ΉΚΆΝΠΚΆΗϋ«ΩΒΡπΛΡΛπΛΗΫΧΊ–‘Θ§ΒΦ÷¬Ρ…ΟΉΝΘΒΡ…χΆΗ–߬ ΒΆ ΘΜΕχΨΜ÷––‘±μΟφΒγΚ…Ω…±ήΟβπΛ“ΚπΛΗΫ [22]ΓΘ»γ”Ο PEG Ε‘ LNΫχ––±μΟφΙΠΡήΜ· ΙΤδ±μΟφ≥ ÷––‘ΚΆ«ΉΥ°–‘Θ§ΡήΩΥΖΰ ηΥ°ΚΆΨ≤ΒγœύΜΞΉς”ΟΕχΆ®ΙΐπΛ“Κά©…Δ [32]ΓΘLN ‘ΎΒΫ¥ο–Γ≥Π ±÷§÷ Ω…±ΜœϊΜ·Υ°Ϋβ≤Δ–Έ≥…¥ΈΦΕΡ“≈ίΘ§Τδ÷§Ϋβ≤ζΈο÷ς“Σ « 2- Η ”ΆΒΞθΞΚΆ÷§ΖΨΥαΘ§»ΜΚσΜα”κΡΎ‘¥–‘Β®÷≠―ΈΒ»œύΜΞΉς”ΟΘ§–Έ≥…≤ΜΆ§ΒΡΫΚ χΈο÷ Θ§–Έ≥…ΒΡΫΚ χ±ΜΉΣ“ΤΒΫΈ¥ΫΝΑηΒΡΥ°≤ψΚΆ…œΤΛΘ§Ά®ΙΐΦρΒΞά©…ΔΒΫ¥ο≥ΠœΗΑϊΒΡΈϋ ’±μΟφΓΘ‘ΎΫΚ χΫαΙΙ÷–»ήΫβΒΡ÷§Ϋβ≤ζΈοΚΆΑϋΙϋΒΡ“©ΈοΩ…¥σΖυ‘ω«ΩΖ÷Ή”‘ΎΈ¥ΫΝΑηΥ°≤ψ÷–ΒΡ¥ΪΒίΘ§¥”Εχ‘ω«Ω“©ΈοΈϋ ’ΓΘ‘ΎœϊΜ·Ιΐ≥Χ÷–Θ§LN ÷§Ϋβ≤ζΈοΚΆ“©ΈοΩ…ΡήΆ®Ιΐ≥ΠΝήΑΆœΒΆ≥ΜρΟ≈Ψ≤¬ω‘Υ δΚΆΈϋ ’ΓΘ
ΧεΡΎΆβ ‘―ι±μΟςΘ§PEG –ό ΈΒΡ SLNs(pSLNs)Ω…―ΗΥΌ¥©ΆΗπΛ“ΚΖ÷ΟΎΈοΘ§ΕχΈό»ΈΚΈ–ό ΈΒΡ SLNs‘ρ“Ή±ΜπΛ“ΚΤΝ’œ≤ΕΜώ [33]ΓΘ“‘–έ–‘ SD ¥σ σΈΣΕ·ΈοΡΘ–ΆΒΡΕύ»α±»–«“©Ε·―ß―–ΨΩ±μΟςΘ§”κ SLNs œύ±»Θ§pSLNs ΨΏ”–ΗϋΗΏΒΡΈϋ ’–߬ ΚΆΗϋ≥ΛΒΡ―Σ“Κ―≠ΜΖ ±ΦδΘ§œύΕ‘…ζΈοάϊ”ΟΕ»ΧαΗΏ 1.99 ±ΕΓΘLN Ω…Ά®ΙΐΕύ÷÷ΖΫ Ϋ”κœΗΑϊœύΜΞΉς”ΟΘ§Αϋά®ΡΎΆΧΉς”ΟΓΔΩγœΗΑϊΉς”ΟΜρ”κœΗΑϊΡΛ»ΎΚœΒ»Θ§¥ΥΆβΜΙΫœ“Ή”κ…χΆΗ¥ΌΫχΦΝΫαΚœΘ§»γΒ®―ΈΚΆ÷§ΖΨΥα’β–©ΒίΥΆœΒΆ≥…θ÷Ν”–Ω…ΡήΗΏ–ßΒΊΩΥΖΰ÷ΈΝΤ–‘ΕύκΡΚΆΒΑΑΉ÷ ΒΡ…œΤΛΤΝ’œ [15]ΓΘ
3.2 LN ΒΡΩΎΖΰΈϋ ’Μζ÷Τ
3.2.1 Ρ…ΟΉ‘Ί“©œΒΆ≥ΒΡΧΊ–‘
ΫϋΡξά¥Θ§Ρ…ΟΉ‘Ί“©œΒΆ≥‘Ύ PPDs ΩΎΖΰΒίΥΆΝλ”ρΒΡ”Π”Ο«ΑΨΑ÷πΫΞΩΣάΪΓΘ Ήœ»Θ§Ρ…ΟΉ‘Ί“©œΒΆ≥ΝΘΨΕΈΣ10 ΓΪ 200 nmΘ§Ω…ΫΪ PPDs »ήΫβΓΔΑϋΙϋΜρΈϋΗΫΤδΦδ“‘Οβ ήΈΗ≥ΠΜΖΨ≥ΤΤΜΒ [34]ΓΘΝΘΨΕ¥σ–ΓΕ‘ΩΎΖΰΡ…ΟΉœΒΆ≥‘ΎΈΗ≥ΠΒάΒΡΈϋ ’”–Ϋœ¥σ”Αœλ ΘΚ≥Ώ¥γΫœ–ΓΒΡΩ≈ΝΘ±»±μΟφΜΐΫœ¥σΘ§ΕχΡ…ΟΉΝΘ‘Ύ…œΤΛœΗΑϊ±μΟφΒΡπΛΗΫ–‘“≤ΥφΉ≈Ω≈ΝΘ±»±μΟφΜΐΒΡ‘ωΦ”Εχ‘ωΦ” [21]Θ§”–άϊ”ΎΧαΗΏPPDs ΒΡ»ήΫβΕ»ΚΆ»ή≥ωΕ»ΓΘ
Τδ¥ΈΘ§Ρ…ΟΉ‘Ί“©œΒΆ≥ΚΆπΛ“Κ÷°Φδ¥φ‘ΎΕύ÷÷œύΜΞΉς”ΟΝΠΘ§”ΑœλΉ≈Ρ…ΟΉ‘ΊΧε‘ΎπΛ“Κ≤ψΒΡά©…ΔΥΌ¬ ΚΆ÷ΆΝτ ±ΦδΓΘπΛ“Κ≤ψΒΡ÷ς“Σ≥…Ζ÷πΛΒΑΑΉ÷ς“Σ”…Χ«ΒΑΑΉΙΙ≥… [3,35]Θ§πΛΒΑΑΉΙ«Φή”…ΥΩΑ±ΥαΓΔΗ§Α±ΥαΚΆΥ’Α±ΥαΒΡ≤–Μυ÷ΊΗ¥–ρΝ–Ήι≥…Θ§”…”ΎΑ±ΜυΥαΒΡΗΏΕ»Χ«ΜυΜ· ΙΤδ‘ωΦ”ΝΥΚήΕύΗΚΒγΚ…Θ§≥ΠΒάπΛ“Κ≤ψ“ρ¥Υ¥χΗΚΒγ [36ΓΣ37]ΓΘπΛΒΑΑΉ”κ‘ΊΧεΒΡΨ≤ΒγΉς”ΟΩ…ΒΦ÷¬ΝΘΉ”ΨέΦ·Θ§¥”Εχ”ΑœλΡ…ΟΉ‘ΊΧε…χΆΗΥΌ¬ ΓΘΕύ ΐ PPDs ΈόΖ®÷ςΕ·ΉΣ‘ΥΘ§Υυ“‘‘ΎΡ…ΟΉ‘ΊΧε±μΟφ…ηΦΤ“Μ≤ψΩΥΖΰπΛΡΛΈϋΗΫΒΡΒγ÷––‘ΜρΒγΗΚ–‘«ΉΥ°≤ψΘ§ΫΪ”–άϊ”ΎΤδ…χΆΗπΛ“Κ≤ψΒΫ¥ο…œΤΛœΗΑϊΡΛ [38]ΓΘ“ΜΑψά¥ΥΒΘ§Ρ…ΟΉ‘Ί“©œΒΆ≥ΒΡ…ψ»ΓΆΨΨΕ÷ς“Σ «Αϊ“ϊ [39]Θ§Μρ M œΗΑϊΆΧ …Ής”ΟΓΘM œΗΑϊΕ‘Ρ…ΟΉ‘Ί“©œΒΆ≥ΒΡ…ψ»ΓΚΆΉΣ‘ΥΟςœ‘ΗΏ”Ύ≥ΠœΗΑϊΘ§≈…ΕϊΑΏ÷– M œΗΑϊΒΡ…ψ»ΓΩ… ΙΡ…ΟΉ‘ΊΧεœΒΆ≥Ά®ΙΐΝήΑΆΈϋ ’Θ§ΫχΕχ»ΤΙΐ ΉΙΐ¥ζ–ΜΒΡ”Αœλ [40ΓΣ41]ΓΘ
3.2.2 ÷§άύΒΡΈϋ ’¥ΌΫχΉς”Ο
÷§άύ”÷≥Τ÷§÷ Θ§ «÷§ΖΨΦΑάύ÷§ΒΡΉή≥ΤΘ§≤Μ»ή”ΎΥ°Εχ“Ή»ή”Ύ÷§ΖΨ»ήΦΝΒ»Ζ«ΦΪ–‘”–Μζ»ήΦΝΘ§≤Δ«“ «ΡήΈΣΜζΧεΥυάϊ”ΟΒΡ÷Ί“Σ”–ΜζΜ·ΚœΈο ΘΜ¥σ÷¬Ω…Ζ÷ΈΣΒΞ¥Ω÷§άύ ( Η ”Ά»ΐθΞ )ΓΔΗ¥Κœ÷§άύ ( ΝΉ÷§ )ΓΔ―ή…ζ÷§άύ ( ÷§ΖΨΥα )ΓΘ÷§άύΨΏ”–ΕάΧΊΒΡ–‘÷ Θ§»γ…ζΈοœύ»ί–‘ΝΦΚΟΓΔΡή‘ω«Ω«Ή÷§–‘Ζ÷Ή”ΒΡΈΗ≥ΠΒάΈϋ ’ΓΔΜ·―ßΫαΙΙΙψΖΚΕύ―υΘ§ Ι÷°≥…ΈΣ”≈ΝΦΒΡΗ≥–ΈΦΝΓΘ‘Ύ≥ΠΒά÷–Θ§÷§άύ ή÷§ΖΨΟΗΒ»”ΑœλΘ§“©Έο¥”÷ΤΦΝ÷– ΆΖ≈≥ωά¥ΚσΜα”κ≥ΠΒά÷–ΒΡΗ ”ΆΕΰθΞΒ»ΖΔ…ζœύΜΞΉς”Ο [42]Θ§÷°ΚσΆ®ΙΐΈϋ ’Β®÷≠―Έ–Έ≥…Έ»Ε®ΫΚ χά¥ Βœ÷‘ω»ήΘ§ΜλΚœΫΚ χ”κ≥ΠœΗΑϊΡΛ…œΒΡ÷§άύΚΆΒΑΑΉ÷ ÷°ΦδΒΡœύΜΞΉς”ΟΘ§ ΙΒΟ÷§÷ ΡΛΗϋΨΏΝςΕ·–‘Θ§‘ω«ΩΝΥ«ΉΥ°–‘ΚΆ«Ή÷§–‘Μ·ΚœΈοΒΡ…χΆΗΡήΝΠΘ§”–άϊ”ΎΫχ“Μ≤ΫΆ®ΙΐΝήΑΆ―≠ΜΖΈϋ ’ [43ΓΣ44]ΓΘΆ§ ±Θ§≥ΠΒάΝήΑΆΉΣ‘Υ–η“ΣΗϋΕύΒΡ ±ΦδΘ§’β“βΈΕΉ≈Ω…ΜώΒΟ“©Έο≥÷–χ ΆΖ≈ΒΡ–ßΙϊ [45]ΓΘ¥ΥΆβΘ§÷§άύΒΡΤδΥϊ”≈ ΤΜΙΑϋά®‘ωΦ”≥ΠœΗΑϊΡΛΆ®ΆΗ–‘ΓΔ±ήΟβ≥ΠΒάΟΗΫΒΫβΦΑΦθ…ΌΗΈ‘ύ ΉΙΐ¥ζ–ΜΓΘ
÷§άύΩ…¥ΌΫχΜν–‘Μ·ΚœΈοΒΡΈϋ ’Θ§‘Ύ…μΧεΈϋ ’÷§ΖΨΒΡΆ§ ±Θ§“©Έο“≤Ηϋ“Ή±Μ…ψ»ΓΘ§’β±Μ»œΈΣ «“Μ÷÷ΓΑΧΊ¬ε“ΝΡΨ¬μΓ±–ß”Π [46]ΓΘΉή÷°Θ§÷§άύ±Μ≥ΠΒά÷–ΒΡΟΗΫΒΫβΘ§‘Ύ÷§ΒΈΜρΙΧΧε÷§ΝΘ±μΟφ–Έ≥…±μΟφΜν–‘ΒΡΒΞΗ θΞΚΆΥΪΗ ”ΆθΞΒ» [32,42]Θ§ Ι÷°‘ΎΒ®Υα―ΈΒ»ΡΎ‘¥–‘Έο÷ Ής”Οœ¬ΨέΦ·–Έ≥…ΫΚ χΘ§‘Ύ¥ΥΙΐ≥Χ÷–Θ§»ήΫβ‘Ύ÷§÷ ÷–ΒΡ“©Έο±ΜΫΚ χ…ψ»ΓΓΔΑϋΙϋΓΘ”…”Ύ÷§άύΒΡΉι≥…ΚΆΧΊ–‘”κœΗΑϊΡΛœύΥΤΘ§“Ή”κ≥ΠπΛΡΛœΗΑϊΖΔ…ζΈϋΗΫΓΔ»ΎΚœΓΔά©…ΔΒ»Ής”ΟΘ§”÷“ρΈΣ LN ΒΡ“©ΈοΑϋΖβΡήΝΠΈ»Ε®Θ§“ρΕχ‘ΎΦ”«Ω“©ΈοΒΡ≥ΠœΗΑϊ…ψ»Γ ±ΜΙΡή Ι“©ΈοΜΚ¬ΐ ΆΖ≈¥”Εχ―”≥ΛΉς”Ο ±Φδ [47]ΓΘ“ρ¥ΥΘ§LN ΒΡ±ΘΜΛΉς”ΟΦ”…œΜΚΩΊ ΆΧΊ–‘Θ§Ω…Ζά÷Ι¥σΖ÷Ή”“©ΈοΙΐ‘γΫΒΫβΘ§≤ΔΧαΗΏΤδΈΗ≥ΠΒάΈ»Ε®–‘ [26]ΓΘ
…œ ω LC ΒΡΩΎΖΰΈϋ ’Μζ÷ΤΉήΫαΦϊΆΦ 1ΓΘ
SLNs ±μΟφΩ…±Μ–ό Έ≥…¥χ“θάκΉ”ΓΔ―τάκΉ”ΜρΨΜ÷––‘±μΟφΒγΚ…Θ§”–―–ΨΩΆ®ΙΐΗζΉΌΤδ‘ΎΧεΡΎΒΡΉΣ‘ΥΘ§ΩΦ≤λΝΥ±μΟφΒγΚ…Ε‘ΩΎΖΰΈϋ ’ΒΡ”Αœλ [48]ΓΘ ‘―ι÷ΛΟς 3÷÷άύ–ΆΒΡ±μΟφΒγΚ…–ό ΈΕΦΡή¥ΌΫχΆξ’ϊ SLNs ΒΡΩΎΖΰΈϋ ’Θ§Τδ÷–ΨΜ÷––‘±μΟφΒγΚ… SLNs ΒΡΈϋ ’ΉνΩλΚΆΉνΕύΘ§’β «“ρΈΣ±μΟφ¥χ÷––‘ΨΜΒγΚ…”κΈ¥–ό ΈΒΡ SLNsΕΦΨΏΫœΒΆΒΡ÷§ΖΨΖ÷ΫβΥΌ¬ ΚΆΫœ«ΩΒΡπΛ“Κ…χΆΗΡήΝΠ( Ω…Ρή «”…”ΎΤδ«ΉΥ°–‘¥ΌΫχΝΥπΛ“Κ…χΆΗ ) ΘΜ“θάκΉ”±μΟφΒγΚ…ΥδΡήΆ®ΙΐΦθΜΚ÷§ΫβΕχ‘ω«ΩΩΎΖΰΈϋ ’Θ§ΒΪΤδ”κ“θάκΉ”œΗΑϊΡΛΒΡœύΜΞΉς”ΟΩ…“ΐΤπΨ≤Βγ≈≈≥β ΘΜ―τάκΉ”ΥδΗϋ ΚœΝΘΉ”ΡΎΜ·»κΡΛΘ§ΒΪ‘ΎΈΗ≥ΠΒά÷–Μα±»÷––‘ΒγΚ… SLN ΗϋΩλΒΊ±Μ÷§ΖΨΟΗΖ÷ΫβΓΘ≤ΜΆ§ΒΡ±μΟφΒγΚ…Ε‘÷§ΫβΒΡ”Αœλ≤Δ≤ΜΖϊΚœΕ‘’ϊΧεΩΎΖΰΈϋ ’ΒΡ”ΑœλΘ§Ω…Ρή‘≠“ρ «¥χΒγΝΘΉ”Μα±ΜΉη÷ΙΫχ»κΧε―≠ΜΖΓΘ’β÷÷¥ΌΫχΩΎΖΰΈϋ ’ΒΡ–ß”ΠΒ±Ιι“ρ”Ύ±μΟφ«ΉΥ°–‘ΒΡ‘ω«ΩΘ§ΕχΖ«±μΟφΒγΚ…ΓΘ ¬ Β…œΘ§Άξ’ϊΩ≈ΝΘΒΡΩΎΖΰΈϋ ’ «Μυ”ΎΦΗΗω“ρΥΊΒΡœύΜΞΉς”ΟΘ§‘Ύ‘ω«ΩΆξ’ϊ SLN ΒΡΩΎΖΰΈϋ ’ΖΫΟφΘ§«ΉΥ°–‘Μρ–μ±»±μΟφΒγΚ…Ηϋ÷Ί“ΣΓΘΆ®Ιΐ±μΟφ–ό ΈΜΙΩ…ΩΊ÷Τ PPDs ΆΖ≈ΒΡΈΜ÷ΟΚΆΥΌ¬ Θ§”–÷ζ”Ύ±ήΟβΖΔ…ζ≤ΜΝΦΖ¥”Π [12,49]ΓΘ
Ήέ…œΥυ ωΘ§ΩΎΖΰ÷§÷ Ρ…ΟΉ‘ΊΧεΒΡ…ηΦΤΡΩ±ξ «Ά®ΙΐΩΊ÷ΤΝΘΨΕ¥σ–ΓΓΔ±μΟφάμΜ·–‘÷ ( ΒγΚ…ΓΔ«ΉΥ° ηΥ°–‘Β» ) ΦΑ÷§÷ ΫαΙΙά¥Ϋχ“Μ≤ΫΧαΗΏ“©ΈοΈ»Ε®–‘ΚΆ ΆΖ≈ΥΌ¬ Θ§¥ΌΫχ“©ΈοΩΎΖΰΈϋ ’Θ§ΧαΗΏ“©ΈοΩΎΖΰ…ζΈοάϊ”ΟΕ» [50]ΓΘ
4 PPDs ΩΎΖΰ÷§÷ Ρ…ΟΉ‘ΊΧεΒΡ÷ς“Σάύ–ΆΦΑΤδ”Π”Ο
4.1 ΙΧΧε÷§÷ Ρ…ΟΉΝΘ (SLNs)
SLNs « 20 άΦΆ 90 Ρξ¥ζ≥θΖΔ’ΙΤπά¥ΒΡΒΎ“Μ¥ζ÷§÷ Ρ…ΟΉΩ≈ΝΘΘ§ «”…ΙΧΧε÷§÷ ΓΔ“©ΈοΚΆ±μΟφΜν–‘ΦΝΑ¥“ΜΕ®±»άΐΉι≥…ΒΡΡ…ΟΉΫΚΧε‘ΊΧε (10 ΓΪ 1 000 nm)[51]ΓΘΙΧΧεΙ«ΦήΧαΙ©ΝΥΗΏΩΙΨέΫαΈ»Ε®–‘ΒΡΩ…Ρή–‘Θ§ΫΒΒΆΝΥ“©ΈοΖ÷Ή”ΒΡΝςΕ·–‘Θ§Ά§ ±Ρή±ΘΜΛΥϋΟ«Οβ ήΜ·―ßΫΒΫβΓΘSLNs ΫαΚœΝΥΨέΚœΈοΡ…ΟΉΝΘΚΆ÷§÷ ΧεΒΡ”≈ΒψΘ§Ά§ ±Φθ…ΌΝΥΈ»Ε®–‘ΦΑΕΨ–‘œύΙΊΈ ΧβΘ§ΜΙΩ…”––ßΩΊ÷Τ“©Έο ΆΖ≈ΥΌ¬ [52]ΓΘ÷§÷ Ρ…ΟΉ÷ΤΦΝ÷–ΒΡ÷§÷ ΜΙΩ…ΡήΜα”ΑœλΥϋΟ«Ά®Ιΐ≥Π…œΤΛœΗΑϊΈϋ ’ΒΡΆΨΨΕ [38]ΓΘ¥ΥΆβΘ§PALIWAL Β»÷Τ±ΗΝΥΦΉΑ±ΒϊΏ SLNsΘ§≤ΔΤάΦέΝΥ÷§άύΕ‘÷ΤΦΝΧΊ–‘ΒΡ”ΑœλΘ§Ιέ≤λΒΫ≤ΜΆ§Ν¥≥ΛΒΡ÷§÷ Ω…ΈΣ“©ΈοΖ÷Ή”ΧαΙ©ΒΡΝ¥Φδ≤ε»κΈΜ÷Ο≤ΜΆ§Θ§¥”Εχ”ΑœλΑϋΖβ¬ [53]ΓΘ≤ΜΆ§–‘÷ ΒΡ÷§÷ Ε‘Η¥»ιΖ®÷Τ±ΗΒΡ SLNs ΒΡ–Έ≥…ΓΔΧεΆβ Ά“©ΚΆΧεΡΎΈϋ ’¥φ‘Ύ”Αœλ [54] ΘΚ»γ÷§÷ »ή“ΚΒΡπΛΕ»Ω…”Αœλ≥§…υΡήΝΩΒΡ¥ΪΒίΘ§¥”Εχ–Έ≥…Η¥»ι“ΚΘΜ÷§άύΚΆ»ήΦΝ÷°ΦδΒΡ ηΥ°–‘≤ν“λΕ‘≥θ»ιΓΔΗ¥»ιΒΡ–Έ≥…ΚΆΉν÷’ΒΡΜ”ΖΔΙΐ≥Χ“≤”–œ‘÷χ”ΑœλΓΘ
CHEN Β»±®Βά”Ο 4 ÷÷÷§÷ [ ”≤÷§Υα (SA)ΓΔ”≤÷§Υα - »ΐΉΊιΒΥαΗ ”ΆθΞ (SA-TP)ΓΔ”≤÷§Υα - »βΕΙόΔΥαΗ ”Ά»ΐθΞ (SA-TM)ΓΔ”≤÷§Υα - ‘¬ΙπΥαΗ ”ΆθΞ(SA-TL)] “‘ΫΚ χ - Η¥»ιΖ®÷Τ±ΗΝΥ 4 ÷÷Αϋ¬ώ SCT ΒΡSLNs[55]ΓΘ―–ΨΩ±μΟςΘ§SA-TP ΒΡΜλΚœΈο‘ΎΗΡ…Τ SLNsΒΡΈ»Ε®–‘ΓΔΧαΗΏ“©Έο‘ΎΡΘΡβ≥Π“Κ÷–ΒΡΈ»Ε®–‘ΦΑ‘ω«ΩSCT ΒΡΈϋ ’ΖΫΟφ–ßΙϊΉνΚΟΓΘΤδœΗΑϊ…ψ»ΓΜζ÷Τ «“άάΒ”ΎΆχΗώΒΑΑΉΚΆ–ΓΈ―ΒΑΑΉΒΡΡΎΆΧΉς”ΟΘ§‘Ύ¥σ σ °Εΰ÷Η≥ΠΡΎΗχ“©Κσœ‘ Ψ≥ωΝΦΚΟΒΡΫΒΗΤΜν–‘Θ§ΩΎΖΰ…ζΈοάϊ”ΟΕ»±» SCT »ή“ΚΗΏ 6 ±ΕΓΘ
Υδ»Μ SLNs Ε‘ PPDs ΨΏ”–Ϋœ«ΩΒΡΈ»Ε®Ής”ΟΘ§ΒΪ“≤ΚήΡ―ΆΜΤΤ≥ΠΒάœΗΑϊπΛ“Κ≤ψΒΡΉηΑ≠Θ§–η“ΣΨ≠Ιΐ Β±ΒΡ±μΟφ–ό Έ≤≈ΡήΖΔΜ”Ής”ΟΓΘΩ«ΨέΧ« «“Μ÷÷Χλ»ΜΒΡ―τάκΉ”ΕύΧ«Θ§“ρΨΏ”–ΝΦΚΟΒΡ…ζΈοœύ»ί–‘ΚΆ…ζΈοΫΒΫβ–‘ΓΔΒΆΕΨ–‘ΓΔΩΙΨζ–‘ΓΔπΛΗΫ–‘ΚΆ‘ω«ΩΈϋ ’ΒΡΧΊ–‘Εχ±Μ”Ο”Ύ“©Έο¥ΪΒίœΒΆ≥ΒΡΩΣΖΔΓΘΨ≠Ω«ΨέΧ«Αϋ±ΜΒΡ÷§÷ Ρ…ΟΉΝΘ”κ»ΥΫα≥ΠœΌΑ©œΗΑϊ (CaCo-2 œΗΑϊ ) ≈ύ―χΡΘ–ΆΒΡœύΜΞΉς”Ο―–ΨΩ±μΟςΘ§ΗΡ…Τ PPDs ΩΎΖΰΈϋ ’ΒΡ‘≠“ρΚήΩ…Ρή «ΗΟœΒΆ≥”κ≥ΠπΛΡΛΖΔ…ζΝΥΝΦΚΟΒΡπΛΗΫΉς”Ο≤Δ‘ω«ΩΝΥ…χΆΗ–‘ [56]ΓΘFONTE Β»≤…”Ο w/o/w –ΆΗ¥»ιΖ®÷Τ±ΗΝΥ¥χΗΚΒγΚ…ΒΡ SLNsΘ§»ΜΚσ”κ¥χ’ΐΒγΚ…ΒΡΒΆΖ÷Ή”Ω«ΨέΧ«ΖΔ…ζΨ≤ΒγœύΜΞΉς”ΟΘ§ΒΟΒΫΩ«ΨέΧ«Αϋ±ΜΒΡSLNs[57]ΓΘ¥σ σΩΎΖΰΩ«ΨέΧ«Αϋ±Μ”κΈ¥Αϋ±ΜΒΡ SLNs ΚσΘ§Ω…Ιέ≤λΒΫ«Α’ΏΫΒΧ«–ßΙϊΟςœ‘ΗΡ…ΤΘ§Ά§ ±Ω…ΦϊΕ®ΈΜ”Ύ≥Π±ΎΦΑΡΎΜ·ΒΫ≥ΠœΗΑϊΡΎΒΡ”ΪΙβ±ξΦ«“»ΒΚΥΊΘ§¥”Εχ÷ΛΟςΝΥΗΟΡ…ΟΉ‘ΊΧε¥ΌΫχ“»ΒΚΥΊ≥ΠΒάΈϋ ’ΒΡ”––ß–‘Θ§“‘ΦΑΑϋ±ΜΩ«ΨέΧ«ΒΡ SLNs Ρή‘ω«Ω≥ΠΒάΕ‘“»ΒΚΥΊΒΡ…ψ»ΓΓΘ¥ΥΆβΘ§Ω«ΨέΧ«ΆΩ≤ψΥΤΚθΡήΫχ“Μ≤Ϋ‘ω«Ω“»ΒΚΥΊΗΚ‘ΊSLNs ΒΡ≥ΠΒάΈϋ ’ΧΊ–‘Θ§’βΩ…Ρή”–÷ζ”ΎΩΣΖΔ“Μ÷÷”≈Μ·ΒΡΩΎΖΰ“»ΒΚΥΊ≈δΖΫ [58]ΓΘ
Ϋχ“Μ≤Ϋ―–ΨΩ±μΟςΘ§Ω«ΨέΧ«Αϋ±ΜΒΡ SLNs ΨΏ”–ΓΑ“ΰ–ΈΓ±ΧΊ–‘Θ§Ω…Χ”±ήΨό …œΗΑϊΒΡΆΧ …ΓΘSARMENTOΒ»“‘ Witepsol( ΑκΚœ≥…“§”ΆθΞ / ΉΊιΒ”ΆθΞ ) ΈΣ÷§ΚΥΘ§≤¥¬ε…≥ΡΖΜρ Tween ΈΣ±μΟφΜν–‘ΦΝ÷Τ±ΗΝΥΤΫΨυΝΘΨΕΈΣ200ΓΪ400 nmΒΡSLNsΓΘΨό …œΗΑϊ…ψ»Γ ‘―ιΫαΙϊ±μΟςΘ§œύΕ‘”ΎΈ¥–ό Έ SLNs ±μœ÷≥ωΒΡΡΎΜ·œ÷œσΘ§Ω«ΨέΧ«ΑϋΙϋΒΡ SLNs Έ¥Φϊ±ΜΡΎΜ·Θ§’β–©ΫαΙϊΈΣ―”≥Λ÷§÷ Ρ…ΟΉΝΘΒΡ―Σ“Κ―≠ΜΖ ±ΦδΧαΙ©ΝΥ–¬ΒΡ―–ΨΩΥΦ¬Ζ [13]ΓΘ
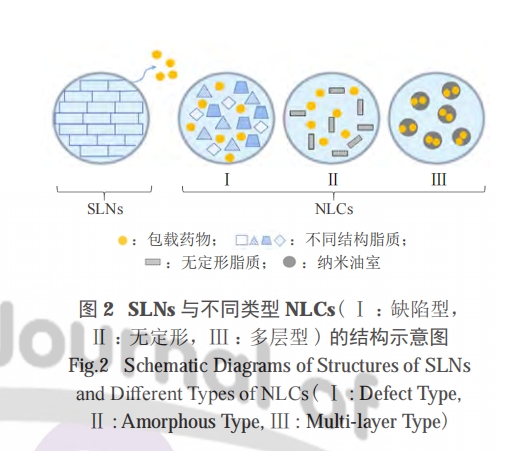
4.2 Ρ…ΟΉΫαΙΙ÷§÷ ‘ΊΧε (NLCs)
NLCs «‘Ύ 21 άΦΆ≥θΖΔ’ΙΤπά¥ΒΡΒΎΕΰ¥ζ÷§÷ Ρ…ΟΉΝΘΘ§”κ SLNs ΒΡ÷ς“Σ«χ±π‘Ύ”ΎΤδΉι≥…÷– Ι”ΟΒΡ÷§άύΒΡΈοάμ–‘÷ ΓΘSLNs Ϋω≤…”ΟΙΧΧε÷§άύΘ§Εχ NLCs≤…”ΟΙΧΧε÷§άύΚΆ“ΚΧ§÷§÷ ( ”Ά ) ΒΡΜλΚœΈοΘ§’βΒΦ÷¬ΝΥ‘ΊΧεΒΡΜυ÷ ΫαΙΙ≤ΜΆ§ΓΘ”κ SLNs œύ±»Θ§NLCs ΒΡ÷ς“Σ”≈ Τ «–μΕύ“©Έο‘Ύ“ΚΧ§÷§÷ ÷–ΒΡ»ήΫβΕ»±»‘ΎΙΧΧ§÷§÷ ÷–¥σΘ§“ΚΧ§÷§÷ ”–÷ζ”ΎΦθ…Ό÷ϋ¥φΤΎΦδΒΡ“©Έο≈≈≥ω [59]ΓΘΗυΨί…ζ≤ζΖΫΖ®ΚΆΜυ÷ ΫαΙΙΒΡ≤ΜΆ§Ήι≥…Θ§Ω…ΜώΒΟ≤ΜΆ§άύ–ΆΒΡNLCsΓΘΔώ–ΆNLCs±Μ≥ΤΈΣ»±œί–ΆΘ§”…ΗΏΕ»Έό–ρΒΡΓΔ≤ΜΆξ…ΤΒΡ÷§÷ Μυ÷ ΫαΙΙΉι≥…Θ§“ρΖ÷Ή”ΦδΒΡΫτΟή≈≈Ν–±Μ¥ρΤΤΘ§Έό–ρΒΡΫαΨßΫαΙΙΩ…»ίΡ…ΗϋΕύΒΡ“©ΈοΖ÷Ή”“‘¥οΒΫ‘ω»ή–ßΙϊΓΘΔρ–Ά NLCs “≤≥ΤΈΣΈόΕ®–ΈΘ§”…ΧΊ βΒΡΈόΕ®–ΈΙΧΧε÷§÷ Μυ÷ ( »γτ«Μυ”≤÷§ΥαθΞ ) ΚΆ“ΚΧ§÷§÷ ΜλΚœΈοΉι≥…Θ§ά以 ±ΡΎ≤Ω÷§÷ “‘ΈόΕ®–ΈΉ¥Χ§ΡΐΙΧ≥…ΈόΕ®–ΈΫαΙΙ‘ΊΧεΒΪ≤ΜΫαΨßΘ§“ρΈΣΫαΨß≥ΘΜαΒΦ÷¬“©ΈοΒΡ≈≈≥ωΓΘΔσ–Ά NLCs ”…ΗΏ≈®Ε»ΒΡ“ΚΧ§÷§÷ Ήι≥…Θ§‘Ύ…ζ≤ζΙΐ≥ΧΒΡά以۹ŸȧΙΧΧε÷§÷ ΚΆ”Ά÷°ΦδΜα≤ζ…ζœύ»ίΦδœΕΘ§ΒΦ÷¬ΙΧΧε÷§÷ Μυ÷ ÷–Ρ…ΟΉΦΕ”Ά«ρΒΡœύΖ÷άκΚΆΈω≥ωΘ§Ήν÷’ΙΧΧεΜυ÷ Αϋ»ΤΉ≈–μΕύ–ΓΒΡ”…“ΚΧ§”Ά–Έ≥…ΒΡΡ…ΟΉ “Θ§“ρ¥Υ≥ΤΈΣΕύ≤ψ–ΆΓΘΗΟΡΘ–ΆΩ…ΩΊ÷Τ“©Έο ΆΖ≈Θ§Εχ÷§÷ Μυ÷ Ω…Ζά÷Ι“©Έο–Ι¬©ΓΘ”…”Ύ“©Έο‘Ύ”Ά÷–ΒΡ»ήΫβ–‘Ά®≥ΘΗΏ”ΎΙΧΧε÷§÷ Θ§“ρ¥ΥΔσ–Ά NLCs Ω… Βœ÷ΗΏ‘Ί“©ΝΩ [60ΓΣ61]ΓΘΜυ”ΎΈοάμΈ»Ε®–‘«ΩΓΔ‘Ί“©ΝΩΗΏΚΆ…ζΈοœύ»ί–‘ΚΟΒ»ΧΊ–‘Θ§NLCs ±Μ»œΈΣ”≈”Ύ¥σ≤ΩΖ÷¥ΪΆ≥ΒΡ LCΓΘ
≤ΜΆ§άύ–Ά NLCs ”κ SLNs ΒΡΫαΙΙ Ψ“βΦϊΆΦ 2ΓΘ
“ρ¥ΥΘ§ΨΓΙή SLNs ΚΆ NLCs ΕΦΩ…ΉςΈΣ PPDs ΒΡ”––ß‘ΊΧεΘ§ΒΪΈ»Ε®–‘ΗϋΚΟΓΔ Ά“©––ΈΣΗϋΦ―ΒΡ NLCs Ω…Ρή «±» SLNs ΗϋΚΟΒΡ―Γ‘ώ [62]ΓΘDUMONT Β»ΩΣΖΔΝΥ“Μ÷÷ΗΏ―ΙΨυ÷ ΦΦ θΫΪ 2 ÷÷«ΉΥ°–‘ΡΘ–ΆΕύκΡ ( »ΞΑ±Φ”―ΙΥΊΚΆΝΝ±ϊ»πΝ÷ ) ΑϋΙϋ‘Ύ”Ο”ΎΩΎΖΰΒΡ NLCs ÷–Θ§2÷÷ΡΘ–ΆκΡΆ®Ιΐ”κΕΰ °ΕΰΧΦΜ«ΥαΡΤΒΡ HIP Ής”Ο–Έ≥…Η¥ΚœΈοΕχ‘ωΦ”Τδ«Ή÷§–‘Θ§ΫαΙϊ÷ΛΟςΥυ÷Τ±ΗΒΡΕύκΡΗ¥ΚœΈοΡή±Μ≥…ΙΠΑϋ¬ώ‘Ύ NLCs ÷–Θ§«“÷ϋ¥φΈ»Ε®–‘ΝΦΚΟ [63]ΓΘ
LC ΉςΈΣ“»ΒΚΥΊΦΑΤδάύΥΤΈοΩΎΖΰΗχ“©‘ΊΧεΒΡΧεΡΎΆβ―–ΨΩΒΟΒΫ≤ΜΕœΆΤΫχΓΘMUNTONI Β»≤…”Ο÷§ΖΨΥαΡΐΨέΖ®÷Τ±ΗΝΥ SLNs ΚΆ NLCsΘ§“»ΒΚΥΊΚΆ“»ΒΚΥΊάύΥΤΈοΗ ΨΪ“»ΒΚΥΊΆ®Ιΐ ηΥ°άκΉ”≈δΕ‘Ής”Ο±ΜΑϋΙϋ‘Ύ÷§÷ Μυ÷ ÷– [26]ΓΘΤδ÷–Θ§ΗΚ‘ΊΗ ΨΪ“»ΒΚΥΊΒΡ NLCs ‘ΎΧεΆβΚΆΧεΡΎ≥ΠΒά±μœ÷≥ωΉνΦ―ΒΡ…ψ»Γ–ßΙϊΘ§ΧεΡΎ“©Ε·―ß―–ΨΩ÷–Ιέ≤λΒΫ―ΣΧ«œ¬ΫΒΘ§’β±μΟς NLCs Ω…“ΜΕ®≥ΧΕ»…œ¥ΌΫχΕύκΡάύ“©ΈοΒΡΩΎΖΰΈϋ ’ΓΘ¥ΥΚσ”÷ΫΪΗ ΨΪ“»ΒΚΥΊ NLCs ΉΣΜ·ΈΣΙΧΧεΩΎΖΰΦΝ–ΆΘ§Ζ÷±πΗχ”ηΫΓΩΒ¥σ σΚΆΧ«Ρρ≤ΓΡΘ–Ά¥σ σΓΘΥφΚσ≤βΕ®ΝΥΗ ΨΪ“»ΒΚΥΊΒΡ…ψ»Γ«ιΩωΚΆΤœΧ―Χ«Ζ¥”Π–‘Θ§Ιέ≤λΒΫ NLCs ΒΡΜλ–ϋ“ΚΚΆΙΧΧε÷ΤΦΝΕ‘ΫΓΩΒ¥σ σΕΦ”–œύ”ΠΒΡΫΒ―ΣΧ«Ής”ΟΘ§ΒΪ÷Μ”–ΫΚΡ“ΦΝΕ‘Χ«Ρρ≤Γ¥σ σ”––ßΘ§÷ΛΟς”… NLCs ΉΣΜ·≥…ΒΡΙΧΧε÷ΤΦΝΨΏ”–ΩΎΖΰΫΒΧ«ΦΝΒΡ”Π”Ο«±ΝΠΘ§”»Τδ «ΫΚΡ“ΦΝ [64]ΓΘ¥ΥΆβΘ§ΆΤ≤βΝΥΫΚΡ“ΦΝΕ‘Χ«Ρρ≤Γ¥σ σ”––ßΒΡ‘≠“ρ ΘΚ ‘―ι÷–Υυ”ΟΒΡΝ¥κεΉτΨζΥΊ (STZ) ”’ΒΦΒΡΧ«Ρρ≤Γ¥σ σ≥ΠΒάπΛΡΛΖΔ…ζΗΡ±δΘ§’β÷÷≤ν“λΒΦ÷¬ΝΥΗ ΨΪ“»ΒΚΥΊΒΡΈϋ ’≤ν“λΘ§Ά§ ±ΫΚΡ“ΒΡΧεΜΐΫœ¥σΘ§ΈΗ≈≈Ω’Φθ¬ΐΘ§”–÷ζ”ΎΗ ΨΪ“»ΒΚΥΊ‘ΎΈΗΚΆ °Εΰ÷Η≥ΠΕΈΒΡΈϋ ’ΓΘSHRESTHA Β»ΤάΙάΝΥ NLCs ΉςΈΣ GLP-1 άύΥΤΈοΓΔΑ§»ϊΡ«κΡΚΆάϊά≠¬≥κΡΒΡΩΎΖΰ‘ΊΧεΒΡ«±ΝΠ [65]ΓΘΫαΙϊœ‘ ΨΘ§NLCs ’Ιœ÷≥ωΕ‘ΕύκΡΒΡΗΏΑϋΖβ¬ Θ§Τδ÷–άϊά≠¬≥κΡΫαΙΙ…œ¥φ‘Ύ“ΜΧθ÷§ΖΨΥαΝ¥Θ§«Ή÷§–‘Ηϋ«ΩΘ§”–÷ζ”ΎΤδΙΧΕ®‘Ύ÷§÷ Μυ÷ …œΘ§“ρ¥ΥΑϋΖβ¬ (95.4ΘΞ )¬‘ΗΏ”ΎΑ§»ϊΡ«κΡ (87.5ΘΞ )ΓΘ”κ”Έάκ“©Έο»ή“Κœύ±»Θ§NLCs ΜΙΡή‘ω«ΩΑ§»ϊΡ«κΡ‘Ύ CaCo-2 ≥ΠΒάΒΞ≤ψ÷–ΒΡ…χΆΗ–‘Θ§»ΜΕχ‘Ύ’ΐ≥Θ–Γ σ÷–ΟΜ”–Ιέ≤λΒΫΟςœ‘ΒΡΫΒ―ΣΧ«–ßΙϊΓΘNLCs ΒΡΫαΙΙ”≈ ΤΜρ–μΨ÷œό”Ύ≥ΠΒάœΗΑϊΒΡπΛ“Κ≤ψΘ§–η“Σάϊ”ΟΗΡ–‘ΦΝΜρΤδΥϊ…ζΈο≤ΡΝœΫχ“Μ≤Ϋ”≈Μ·ΓΘ
5 Ϋα¬έ”κ’ΙΆϊ
ΒΑΑΉΕύκΡΒ»…ζΈο¥σΖ÷Ή”Ε‘Α–ΒψΒΡ―Γ‘ώ–‘«ΩΓΔΜν–‘ΗΏΓΔΝΤ–ßœ‘÷χΘ§Ρή‘ΎΧ«Ρρ≤ΓΓΔ÷ΉΝωΒ»Φ≤≤Γ÷ΈΝΤ÷–ΖΔΜ”÷Ί“ΣΉς”ΟΓΘ»ΜΕχΘ§¥σΕύ ΐ…ζΈο¥σΖ÷Ή”“©ΈοΩΎΖΰΚσ±ΜœϊΜ·ΟΗΫΒΫβΘ§«“«ΉΥ°–‘«ΩΓΔΡ―“‘ΆΗΡΛΘ§ΒΦ÷¬ΩΎΖΰ…ζΈοάϊ”ΟΕ»ΒΆΓΘΥδ»ΜΕ‘ PPDs Ϋχ––Μ·―ß–ό ΈΚΆΦ”»κΗΡ–‘ΦΝΩ…ΧαΗΏΈ»Ε®–‘ΓΔ‘ωΦ”ΡΛΆ®ΆΗ–‘ΓΔ¥ΌΫχ–Γ≥ΠΈϋ ’Θ§ΒΪ“≤Ω…Ρή‘ω«ΩΈΗ≥ΠΒάΕΨ–‘ΓΘπΛΡΛπΛΗΫœΒΆ≥Ρή―”≥Λ PPDs ‘ΎΈΗ≥ΠΒάΒΡ÷ΆΝτ ±ΦδΘ§ΧαΗΏ…ζΈοάϊ”ΟΕ»Θ§ΒΪ»¥≤ΜΡήΧαΗΏ“©ΈοΒΡΩΎΖΰ…χΆΗ–‘Θ§“≤≤ΜΡή±ήΟβ–Γ≥ΠπΛΡΛΒΡ«ε≥ΐΉς”ΟΓΘΕχ÷§÷ Ρ…ΟΉΒίΥΆ‘ΊΧε (SLNsΚΆ NLCs Β» ) ΨΏ”–ΤδΥϊΖΫΖ®ΈόΖ®±»ΡβΒΡ”≈ ΤΘ§”Β”–ΝΦΚΟΒΡΈ»Ε®–‘ΚΆ…ζΈοœύ»ί–‘Θ§Ω…±ΘΜΛΑϋ‘Ί“©Έο≤Μ ήΟΗΫΒΫβΚΆ pH ΧθΦΰΒΡ”ΑœλΘ§άϊ”ΟπΛΗΫΧΊ–‘ΫΪΡ…ΟΉΝΘ…χΆΗ÷Ν…œΤΛœΗΑϊΡΛ…œΘ§άϊ”ΟΝήΑΆΈϋ ’ΆΨΨΕ”–÷ζ”Ύ“©Έο±ήΟβ ΉΙΐ¥ζ–ΜΘ§Ηϋ÷Ί“ΣΒΡ «“―±Μ≥…ΙΠΒΊ”Π”Ο”ΎPPDs ΩΎΖΰΗχ“©Θ§»γ“»ΒΚΥΊΚΆΫΒΗΤΥΊΓΘ
÷ΒΒΟ“ΜΧαΒΡ «Θ§SLNs ΚΆ NLCs Ε‘«ΉΥ°–‘“©ΈοΒΡ‘Ί“©ΝΩ”–œόΘ§―–ΨΩΖΔœ÷Ω…Ά®ΙΐΙ≤ΦέΦϋΜρΖ«Ι≤ΦέΦϋ–Έ ΫΫΪ PPDs ”κ÷§άύΜ·ΚœΈο≈ΦΝΣΘ§ΉΣΜ·ΈΣΗϋ«Ή”ΆΓΔ≤Μ»ή”ΎΥ°ΒΡΖ÷Ή”Θ§“‘¥¥‘λΗΚ‘Ί–߬ ΗΏΒΡ÷§÷ Ρ…ΟΉΒίΥΆœΒΆ≥ [34]ΓΘΆ§ ±÷§÷ Ρ…ΟΉΝΘ±μΟφΜΙΩ…–ό ΈœΗΑϊ¥©ΆΗκΡΜρ«ΉΥ°–‘ΨέΚœΈοΒ»Θ§ΖΔΜ”Εΰ’Ώ‘ΎΩΎΖΰΗχ“©ΖΫΟφΒΡ–≠Ά§”≈ ΤΘ§ΈΣ PPDs ΩΎΖΰΒίΥΆΧαΙ©«±‘Ύ≤Ώ¬‘ΓΘSLNsΓΔNLCs ΦΦ θ“―‘Ύ»Ϊ«ρΜ·Ή±ΤΖΚΆ“©Ή±≤ζΤΖ –≥Γ÷–ΒΟΒΫ”Π”ΟΘ§ΒΪΒΫΡΩ«ΑΈΣ÷ΙΘ§”Ο”Ύ PPDs ÷ΈΝΤ”ΟΆΨΒΡ SLNs ÷ΤΦΝΜΙ¥Π”ΎΝΌ¥≤ ‘―ιΫΉΕΈΘ§NLCs ÷ΤΦΝ“≤÷Μ¥Π”Ύ Β―ι “ΩΣΖΔΫΉΕΈΓΘΈΣ≥δΖ÷ΖΔΨρ÷§÷ Ρ…ΟΉΒίΥΆ‘ΊΧεΉςΈΣ PPDs ΩΎΖΰœΒΆ≥ΒΡ«±ΝΠΘ§–η“ΣΦ”ΩλΝΌ¥≤«ΑΚΆΝΌ¥≤…œΒΡ―ι÷Λ―–ΨΩΘ§Ά§ ±Ϋ®ΝΔΉΦ»ΖΒΡΧεΡΎΆβœύΙΊ–‘ΡΘ–Ά“≤Ε‘¥ΌΫχΤδΝΌ¥≤ΉΣΜ·ΨΏ”–÷Ί“Σ“β“ε [66]ΓΘ¥ΥΆβΘ§…ν»κΝΥΫβ÷§÷ Ρ…ΟΉΒίΥΆ‘ΊΧεΒΡΧεΡΎΈϋ ’Μζ÷ΤΦΑΖ÷≤ΦΫΪ”–÷ζ”Ύ…ηΦΤΗϋ”––ßΒΡΒίΥΆœΒΆ≥Θ§œύ–≈ΥφΉ≈ΒΑΑΉ÷ Μ·―ßΓΔ÷§÷ Ρ…ΟΉ÷ΤΦΝΚΆ“©ΈοΒίΥΆΒΡΦΦ θ―ΗΥΌΖΔ’ΙΘ§Μυ”Ύ÷§÷ ΒΡΡ…ΟΉ‘ΊΧεΫΪΫχ“Μ≤ΫΥή‘λ PPDs ΩΎΖΰΒίΥΆΒΡ–¬ΗώΨ÷ΓΘ
Οβ‘π…υΟςΘΚ±ΨΈΡΈΣ––“ΒΫΜΝς―ßœΑΘ§Αφ»®Ιι‘≠Ής’ΏΦΑ‘≠‘”÷ΨΥυ”–Θ§»γ”–«÷»®Θ§Ω…ΝΣœΒ…Ψ≥ΐΓΘΈΡ’¬±ξΉΔ”–Ής’ΏΦΑΈΡ’¬≥ω¥ΠΘ§»γ–η‘ΡΕΝ‘≠ΈΡΦΑ≤ΈΩΦΈΡœΉΘ§Ω…‘ΡΕΝ‘≠‘”÷ΨΓΘ
