
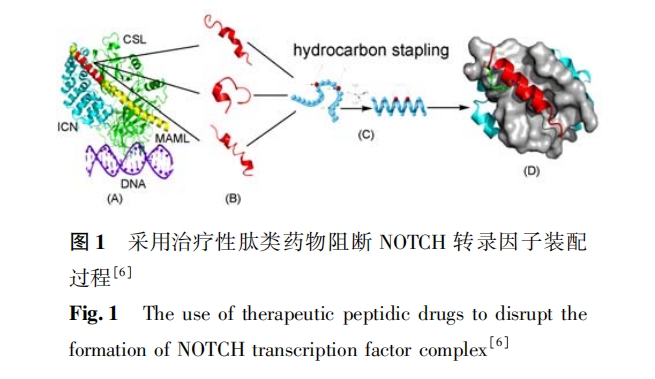
еЊ вЊ ыФзїЮЊживЊЕФЩњРэЛюадЮяжЪвЛжБЪмЕНЯрЙиСьгђЕФЙуЗКЙизЂЁЃ НќФъРДЃЌгЩгкыФдкЯИАћаХКХзЊЕМжаЫљАчбнЕФжааФНЧЩЋвдМАзїЮЊЩњЮявЉЮяАаЯђЕААзжЪЯрЛЅзїгУЭјТчЕШЬиЪтаджЪЕФЗЂЯжЃЌдйДЮЛНЦ№СЫШЫУЧЖдыФЕФХЈКёаЫШЄЁЃ гыжЎЯрАщЕФЪЧЃЌыФЕФРэТлКЭМЦЫубаОПЙЄзїПьЫйдіГЄЃЌВЂШЁЕУСЫГЄзуНјеЙЁЃБОЮФвд"МЦЫуыФбЇ"ЮЊжїЬтЯЕЭГИХРЈСЫИУСьгђЕФбаОПЗЖГыКЭбаОПЬиЕуЃЌВЂЗжБ№ДгыФЕФЪ§ОнПтЙЙНЈЁЂЙІФмЛюаддЄВтЁЂЗжзгЖдНгЁЂЖЏСІбЇФЃФтЁЂНсЙЙЪ§ОнЗжЮіЁЂЗжзгЩшМЦаоЪЮвдМАЯЕЭГЩњЮябЇааЮЊЕШМИЗНУцЗжРрНщЩмСЫМЦЫуыФбЇЕФжївЊбаОПЗНЯђКЭЕБЧАЗЂеЙзДПіЁЃжиЕудкгкЬНЬжВЩгУМЦЫуЛЏбЇКЭЩњЮяаХЯЂбЇЗНЗЈЦЪЮіыФгыЕААзжЪЪЖБ№КЭЯрЛЅзїгУЕФЗжзгЛњжЦКЭРэЛЏЛљДЁЃЌНјЖјЮЊыФРрвЉЮяЩшМЦЬсЙЉРэТлжИЕМЁЃДЫЭтЃЌБОЮФЛЙЬсГіСЫМЦЫуыФбЇдкыФРрФЩУзВФСЯМАЩњЮяБэУцЛюадМСЕШСьгђЕФЧБдкгІгУЧАОАЁЃ
ыФЪЧвЛжжживЊЕФФкдДЩњРэЛюадЮяжЪЃЌЫќУЧзїЮЊЗжУкМЄЫиЁЂЩёОЕнжЪЁЂПЙдБэЮЛЕШВЮгыСЫЛњЬхЩњГЄЗЂг§ИїИіНзЖЮЕФИДдгЩњРэЛюЖЏКЭЯИАћЙ§ГЬЁЃНќФъРДЫцзХЗжзгЩњЮябЇКЭЕААзжЪзщбЇЕФПьЫйЗЂеЙЃЌЁАыФЁБетИіИХФюЕУЕНСЫМЋДѓЕФГфЪЕКЭРЉеЙЃКЯжДњыФПЦбЇЕФбаОПЗЖГыВЛдйОжЯогкЙТСЂЕФЙбОлАБЛљЫсађСаЃЌвВАќРЈФЧаЉвдЯпадФЃЬхаЮЪНЗЂЛгЩњЮябЇЙІФмЕФЕААзжЪФкЧЖШсадЦЌЖЮЁЃЩѕжСЛЙКИЧвЛВПЗждкЕААзжЪБэУцГЩЭХЗжВМЕЋвЛМЖађСаВЂВЛНгСЌЕФЙІФмадВаЛљЃЈШчBЯИАћБэЮЛЃЉЁЃгЩДЫЃЌвтДѓРћАЭРяДѓбЇЕФ Lucchese ЕШТЪЯШЬсГіыФбЇЃЈ peptidologyЃЉетИізЈУХИХ ФюгУгкЗКЛЏЯжДњыФПЦбЇЕФбаОПЗЖГы [3]
ЪТЪЕЩЯ ,зюНќСНЗНУцПЦбЇНјеЙЮЊыФбЇСьгђзЂШыСЫОоДѓЛюСІ ЁЃвЛЗНУц ,ШЫУЧж№НЅШЯЪЖЕНДцдкгкецКЫ ЛљвђзщжаДѓСПДІгкНсЙЙЮоЖЈаЮЬЌЕФЕААзжЪМАЕААзЦЌЖЮЃЈ intrinsically disordered proteinЃЉВЂЗЧШчЯШЧАШЯЮЊЕФФЧбљЪЧУЛгаЙІФмЕФЁА ЩњЮяЗЯСЯЁБ,ЫќУЧПЩвдЭЈЙ§НсКЯ-елЕўХМКЯЃЈ folding-on-bindingЃЉЗНЪНВЮгыЕААзжЪЪЖБ№ВЂећКЯШыЯИАћаХКХЭЈТЗЕБжа [4] ЁЃдкИУЙ§ГЬжа ,ДЫРрЮоЖЈаЮЕААзвдЦфБэУцвЛЖЮШсадЖЬыФЦЌЖЮгыЯТгЮЕААзЕФИеадыФЪЖБ№гђЃЈ peptide-recognition domainЃЉЗЂЩњЪЖБ№КЭНсКЯ ,ДгЖјНщЕМФИЬхЕААзжЎМфЖЬднЖјПЩФцЕФЯрЛЅзїгУЃЛСэвЛЗНУц , гЩгкЕААзжЪЭјТчЯрЙиМВВЁзщЕФзюаТбаОПНјеЙ ,ЪЙЕУвдЕААзжЪЯрЛЅзїгУЮЊАаБъЕФаТвЉбаЗЂв§Ц№ЯрЙиСьгђбаОПепЕФЙуЗКаЫШЄЁЃ ШЛЖј , аэЖрЕААзжЪЯрЛЅзїгУвдаЮГЩПэЗКЖјЦНЬЙЕФЪшЫЎНчУцЮЊЬиеїЃЈ ШчзЊТМвђзгИДКЯЮяЃЉ,ДЋЭГаЁЗжзгЛЏбЇвЉЮя ЖдЦфаЇЙћВЛМб [5] ЁЃ ШЁЖјДњжЎЕФЪЧВЩгУыФРрЗжзгЪЕЬхЃЈ peptide entityЃЉФЃФтИДКЯЮяНчУцЕФЙиМќГЩЗж ,ДгЖјДяЕНОКељадзшЖЯИУРрзїгУЕФФПЕФЃЈ ЭМ 1ЃЉЁЃ
МјгквдЩЯЪТЪЕ , НќФъРДеыЖдыФЕФбаОПГЩЮЊЁА ЗКЗжзгЁБПЦбЇМвЙВЭЌЙизЂЕФНЙЕу ЁЃ ЕБЧА ,баОПепВЛдйНЋыФНіОжЯоЮЊЩњЮяЛюадЮяжЪ ,ЖјЧввбНЋЦфПЊЗЂЮЊЙІФмФЩУзВФСЯ [7] ЁЂЩњЮяБэУцЛюадМС [8] КЭвНгУЗжзгЬНеы[9] ЕШЖржжгУЭОЁЃЫцзХыФбЇЬхЯЕЕФж№ВНЭъЩЦ ,еыЖдыФМАЦфРрЫЦЮяПЊеЙЕФРэТлКЭМЦЫубаОПГЪЯжГіЗНаЫЮД АЌЕФЧїЪЦЁЃетаЉбаОПМИКѕЩцМАЕНСЫЯжДњМЦЫуЛЏбЇКЭЩњЮяаХЯЂбЇЕФЗНЗНУцУц ,ЦфАќРЈЕЋВЛНіЯогкЗжзгЖЏСІбЇФЃФтЁЂСПзгСІбЇБэеїЁЂЭГМЦЭЦЖЯдЄВтЁЂОЇЬхНсЙЙЦЪЮіЁЂзщбЇЭјТчЙЙдьЁЂЪ§ОнзЪдДЗЂОђЁЂЛљгкНсЙЙЕФХфЛљЩшМЦЕШЁЃдчЦк ,ЮвУЧПЮЬтзщврдкДЫСьгђПЊеЙСЫвЛ ЯЕСаЬНЫїЙЄзї [10ЁЊ21] ЃЛдкИУЙ§ГЬжа ,ЮвУЧгњЗЂОѕЕУгаБивЊНЋжЎЩЯЩ§ЮЊвЛУХЬиЖЈбЇПЦМгвдЯЕЭГИХРЈЁЃМјгкДЫЯШ Lucchese ЕШвбВЩгУЁАыФбЇЃЈ peptidologyЃЉЁБРДУшЪіыФЯрЙиПЦбЇЕФбаОПЗЖГы [3]ЁЃЮЊСЫЗНБуЦ№Мћ ,врРрЫЦ гкЁАМЦЫуУИбЇЃЈ computational enzymologyЃЉЁБ[22] ,етРя ГЂЪдвдЁАМЦЫуыФбЇЃЈ computational peptidologyЃЉЁБетИі ЬсЗЈРДИВИЧИУСьгђЁЃЭМ 2 ИјГіЕБЧАМЦЫуыФбЇжївЊба ОПЗНЯђЕФЪїзДДгЪєЙиЯЕЁЃашвЊЫЕУїЕФЪЧДЫЪїЭМЕФЙЙдьГігкжїЙл ,ЦфИїИіЗжжЇОпгаНЯДѓЕФжиЕўадКЭНЛВцадЃЛетбљЛЎЗжжївЊЮЊСЫБугкЯТЮФЗжРрЬНЬжЁЃ
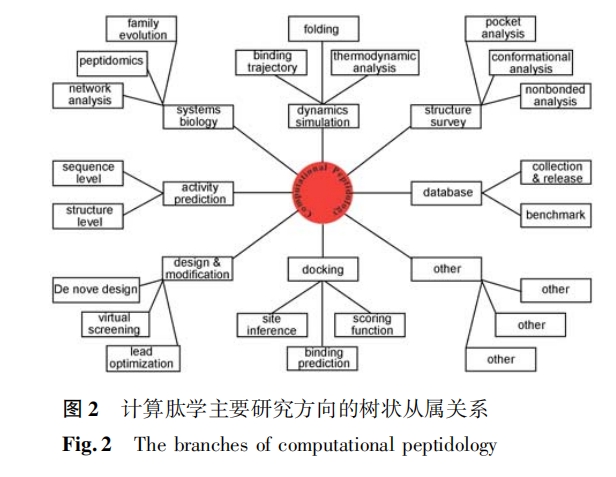
БОзлЪіЗжЮЊ 3 ИіВПЗж ,ЦфФкШнАВХХШчЯТЃКЃЈ1ЃЉв§ бдВПЗжНщЩмыФбЇЕЎЩњЕФбЇПЦБГОАвдМАдкДЫБГОАжааЮГЩЕФМЦЫуыФбЇетИіЗжжЇСьгђ ,зїЮЊЬсв§ЃЛЃЈ2ЃЉбаОПЗНЯђВПЗжЗжРрЬНЬжМЦЫуыФбЇЕФбаОПФкШнКЭзюаТНјеЙ , зїЮЊжїЬхЃЛЃЈ3ЃЉзмНсМАеЙЭћВПЗжеыЖдМЦЫуыФбЇЕФЕБЧАбаОПзДПіМАЮДРДЗЂеЙЧїЪЦМгвдМђвЊИХРЈ , зїЮЊЪеЮВ
2 МЦЫуыФбЇЕФбаОПЗНЯђ
2. 1 ыФЕФЪ§ОнПтЙЙНЈ
ЦљНёПЦбЇЙВЭЌЬхзмЙВЗЂВМСЫМИЪЎИіыФЯрЙиЪ§ОнПт ,ЦфжаЖрЪ§ЪЧеыЖдЬиЖЈФПЕФНЈСЂЦ№РДЕФыФађСа-ЙІФмПт ,ШчУтвпБэЮЛПт IEDB[23] ЁЂПЙОњыФПт APD[24] ЁЂЙІФмЪГЦЗыФПт BIOPEP[25] вдМАзлКЯадЕФЛюадыФЪ§ОнПт PepBank [26] КЭ BioPD[27] ЁЃетаЉзЪдДЖдгкбаОПыФЕФвЛ МЖађСаФЃЪНгыЬиЖЈЛюадЙІФмЙиЯЕДјРДСЫМЋДѓБуРћ , вВЮЊЩњЮяаХЯЂбЇМвЗЂеЙЛњЦїбЇЯАФЃаЭЬсЙЉСЫЗсИЛЕФЪ§ОнРДдД ЁЃШч IEDB Ъ§ОнПтвбГЩЮЊЕБЧАУтвпаХЯЂбЇбаОПЕФЙЋЙВЪ§ОнЦНЬЈ ,ШЫУЧРћгУЦфЙуЗКПЊеЙСЫПЙдыФЗжЮіКЭыФвпУчЩшМЦЕШЗНУцЕФбаОПЁЃ
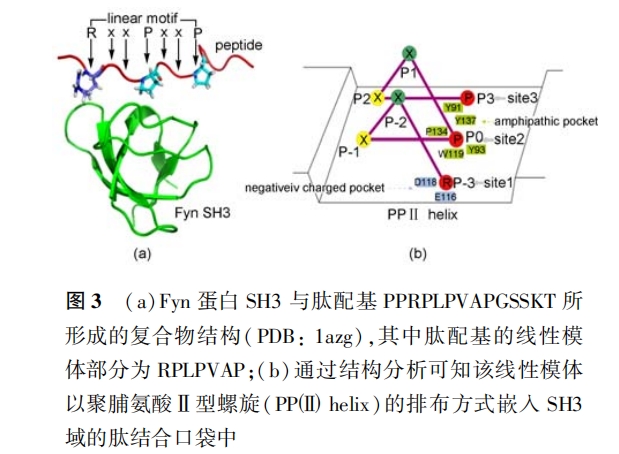
НќФъРД ,АќКЌЕААзжЪ/ыФЪЖБ№КЭЯрЛЅзїгУаХЯЂЕФ Ъ§ОнПтдкНЯИпВуДЮзЂЪЭСЫыФааЪЙЦфЩњЮяЙІФмЕФЗжзгЛњжЦ , вђЖјГЩЮЊаТвЛДњыФЪ§ОнПтбаЗЂЕФЧїЪЦЁЃASPD ЪЧдчЦкЗЂВМЕФвЛИізЈУХЪеМЏКЭзЂЪЭыФЬхЭтЖЈЯђНјЛЏЪЕбщЕФЪ§ОнПт[28] ,жЎКѓ DOMINO ећКЯСЫЪ§ЧЇжжгђ/ ыФЯрЛЅзїгУаХЯЂ [29] ЁЃДЫЭт ,БЪепЫљдкЭХЖгзюНќПЊЗЂЕФФЃФтБэЮЛПт MimoDB ЯЕЭГзЂЪЭСЫЩЯЭђЬѕЪЩОњЬхеЙЪОФЃФтыФЕФЯрЙиаХЯЂКЭЖдгІЪмЬхЗжРр [30] ЁЃгЩгкЕААзжЪЃЈЛђыФЪЖБ№гђЃЉЭљЭљЭЈЙ§ЪЖБ№ОпгаЬиЖЈађСаФЃЪНЕФЙбыФЦЌЖЮРДгыжЎНсКЯ ,етаЉађСаФЃЪНГЦЮЊЖЬЕФЯпадФЃЬхЃЈshort linear motif, SLiMЃЉЁЃШч SH3 гђЬивьадЪЖБ№Опга + xxPxxP Лђ PxxPx + ФЃЪНЕФОлИЌАБЫсM аЭТна§ыФЃЈЗћКХАДее EURESCO ЛсвщЭЦМі[31] ЃЛетРя x ЮЊШЮвтВаЛљ, + ЮЊжЪзгЛЏе§ЕчКЩВаЛљ,P ЮЊИЌАБЫсЃЉ ЃЈЭМ 3ЃЉЃЛСэЭтСНРрГЃМћЕФыФЪЖБ№гђЪЧ SH2 КЭ PDZ,ЧАепзЈЫОЪЖБ№ДјИКЕчКЩЕФСзЫсЛЏыФЦЌЖЮ ,ЖјКѓепдђНсКЯЕААзжЪєШЛљЖЫЕФЙбыФађСа ЁЃвђДЫЪеМЏЁЂзЂЪЭКЭЗжРр SLiM ЕФзЈУХадЪ§ОнПтврЮЊбаОПЕААзжЪ/ыФЯрЛЅзїгУЕФживЊЙЄОп , ЦфДњБэАќРЈ ELM[32] ЁЂMnM [33] КЭ PROSITE [34] ЕШЁЃ
ИќИпвЛИіВуДЮЕФдзгНсЙЙЫЎЦН , ФПЧАжївЊДцдкСНИізлКЯадЕФЕААзжЪ/ыФИДКЯЮяЪ§ОнПт , МД 3did[35] КЭ Pepx[36] ЃЛЫќУЧДгЩњЮяДѓЗжзгНсЙЙЪ§ОнПт PDB[37] жаЬсШЁЕААзжЪЃЈЛђНсЙЙгђЃЉгыыФаЮГЩИДКЯЮяЕФПеМфНсЙЙЪ§ОнВЂМгвдЙщРрзЂЪЭ ,ШчЭЈЙ§ЖдБШЕААзжЪБэУцыФНсКЯПкДќЕФПеМфЙЙдьЖјЖдИДКЯЮяНјааЗжРр ЁЃетРрЪ§ОнПтвбБЛЙуЗКгУгкЕААзжЪ/ыФЯрЛЅзїгУНсЙЙаХЯЂ ЗжЮіКЭЬсШЁЃЈВЮМћ 2. 5 НкЃЉЁЃвХКЖЕФЪЧ ,етаЉНсЙЙЪ§ОнПтВЂЮДЬсЙЉЯргІЛюадЛђЧзКЯСІаХЯЂ ЁЃвЛАуШЯЮЊ , вЊЯыШЋУцЩюШыбаОПЩњЮяЗжзгЪЖБ№ЛњжЦ , Г§СЫЛёжЊИДКЯЮяЕФШ§ЮЌНсЙЙвдЭтЛЙашСЫНтИУНсЙЙЖдгІЕФЩњЮяЛюаджЕ ЁЃвђДЫ ,ПЩвддЄСЯ ,ЙЙНЈНсЙЙ-ЛюадЙиСЊЪ§ОнПтНЋГЩЮЊыФЪ§ОнПтЮДРДЗЂеЙЗНЯђжЎвЛЁЃ
2. 2 ыФЕФЙІФмЪЖБ№МАЛюаддЄВт
ОЋШЗПЩППЕФМЦЫуЪЖБ№КЭдЄВтЩњЮяЛюадВЛНіЪЧЖдыФЖјЧвЖдЦфЫћЩњЮяЗжзгЖјбдЖМЪЧжСЙиживЊЕФ , вђЮЊетЪЧИпЭЈСПащФтЩИбЁКЭРэадЗжзгЩшМЦЕФЛљДЁ ЁЃФПЧА ,ыФЕФЙІФмЪЖБ№МАЛюаддЄВтжївЊдкЛљгкађСаКЭЛљгкНсЙЙСНИіЗНУцеЙПЊ ЁЃЧАепНіРћгУыФЕФвЛМЖађСааХЯЂ ,ВЩгУЭГМЦНЈФЃЗНЗЈ ,гыЯргІЩњЮяЛюадМгвдКЏЪ§Йи СЊ ,НЈСЂдЄВтФЃаЭЃЛКѓепдђЭкОђвбжЊЕФЕААзжЪ/ыФИДКЯЮяНсЙЙаХЯЂ , ВЂДгПеМфдзгВуДЮдЄВтыФЕФЩњЮяЛюадЁЃ
ЃЈ1ЃЉ ађСаВуДЮ : дкЖЈадЫЎЦН ,ВЩгУШевцЗсИЛЕФ SLiM жЊЪЖЭЦВтЕААзжЪЙІФмЮЛЕуГЩЮЊГЃгУВпТдЁЃИУЗЈгЩгкЦфМђЕЅИпаЇЖјБЛЙуЗКгУгкдкЛљвђзщЫЎЦНдЄВт аХКХыФЁЂУИЧаЮЛЕуЁЂСзЫсЛЏЧјгђКЭыФХфЛљ [38] ЁЃИќНјвЛВН ,ЩњЮяаХЯЂбЇМвЪЙгУДѓСПвбжЊЙІФмыФађСабЕСЗЛњЦїбЇЯАЙЄОпЃЈ ШчШЫЙЄЩёОЭјТчКЭжЇГжЯђСПЛњЃЉ,МЬЖјгУгкЦРЙРЮДжЊбљБО [39] ЁЃдкЖЈСПЫЎЦН ,ЛЏбЇМЦСПбЇМвЗЂеЙЦ№РДЕФЖЈСПЙЙаЇЙиЯЕЃЈQSARЃЉГЩЮЊдЄВтыФЩњЮяЛюадЕФГЃМћЪжЖЮ[40] ЁЃHellberg ЕШдчЦкЕФбаОПЕьЖЈСЫИУЗЈЛљБОПђМм ,ЦфЫМТЗЮЊ [41] : ЪеМЏАБЛљЫсЕФДѓСПаджЪВЮЪ§ЃЛВЩгУжїГЩЗжЗжЮіЃЈPCAЃЉЬсШЁетаЉВЮЪ§жаЫљвўКЌЕФЯджјаХЯЂЕУЗж , ГЦЮЊжїаджЪЃЈprincipal propertiesЃЉЃЛЪЙгУетаЉЩйСПжїаджЪДњЬцДѓСПдЪМБфСП , Лё ЕУ Ыљ ЮН ЕФ АБ Лљ Ыс Уш Ъі згЃЈ amino acid descriptorsЃЉПЩгУвдВЮЪ§ЛЏыФЕФвЛМЖађСаНсЙЙЃЛНјЖј ВЩгУЦЋзюаЁЖўГЫЃЈ PLSЃЉЕШЛиЙщММЪѕЭГМЦЙиСЊыФЕФНсЙЙВЮЪ§гыЩњЮяЛюад ,ЛёЕУЕФКЏЪ§ЙиЯЕПЩгУгкдЄВтФПЕФЁЃ ГЃМћАБЛљЫсУшЪізгАќРЈ Z БъЖШ[41] ЁЂISA-ECI жИ Ъ§ [42] ЁЂVHSE ЕУЗж[43] ЕШЁЃИУЗЈЕФШБЕуЪЧЮоЗЈДІРэГЄЖШВЛвЛЕФыФађСаЁЃЫфШЛКѓРДгаШЫЬсГіСЫжюШчздНЛВцаЗНВюЃЈ auto-cross covariance, ACCЃЉ[44] ЕШЗНЗЈМгвдНтОі ,ЕЋвђЮяРэвтвхВЛЩѕУїШЗЖјЮДЕУЕНЙуЗКгІгУЁЃ
ЃЈ2ЃЉНсЙЙВуДЮ : гЩгкНсЙЙЪ§ОнДѓДѓЩйгкађСаЪ§Он , вђДЫЛљгкНсЙЙдЄВтыФЕФЩњЮяЛюадЛЙЯЪМћБЈЕР ,ЕЋЫќОпгааэЖрЯрЖдгкЛљгкађСадЄВтЗНЗЈЫљВЛПЩБШФтЕФ гХЪЦ ,ШчНсЙћЮяРэЛЏбЇвтвхУїШЗЁЂЫљЛёжЊЪЖвзгкжИЕМНсЙЙИФдьЕШЁЃвдЭљ ,ЛљгкНсЙЙдЄВтыФЛюадбаОПзюЖрЕФЖдЯѓЪЧжївЊзщжЏЯрШнадИДКЯЮяЃЈMHCЃЉ[45] , ЫќЪЧживЊЕФЯИАћУтвпЯрЙиЕААз ,зЈЫОИКд№АћФкыФПЙдМгЙЄЬсГЪ ,ЖдвпУчЩшМЦвтвхжиДѓЁЃвђДЫЯжгаДѓСПОЇЬхНсЙЙЪ§ОнПЩЙЉРћгУ [46] ЁЃСэЭт ,гђ/ыФЯрЛЅзїгУвВЪЧ ЛљгкНсЙЙдЄВтыФЛюадЕФЙизЂШШЕу ,жївЊбаОПЖдЯѓАќ РЈ SH3 гђЁЂWW гђЁЂPDZ гђЕШЁЃШч Hou ЕШВЩгУЭЌдДФЃНЈЁЂЗжзгЖдНгМАЖЏСІбЇФЃФтдкНсЙЙЫЎЦННвЪОСЫШЫРрЫЋдиЕААз SH3 гђЃЈ hAmph SH3 ЃЉгыХфЛљыФЕФзїгУФЃЪН ,ВЂдкДЫЛљДЁЩЯНЈСЂСЫгУгкыФЧзКЯСІдЄВтЕФШ§ЮЌЖЈСПЙЙаЇЙиЯЕЃЈ3D-QSARЃЉФЃаЭ[47] ЁЃ жЎКѓЫћУЧгжЬсГіЗжзгЯрЛЅзїгУФмЗжНтгыжЇГжЯђСПЛњСЊгУЗНАИ ЃЈMIEC-SVM ЃЉЩИ бЁ Лљ вђ зщ жа ЕФ ЧБ дк SH3 ыФ Хф Лљ [48,49] ,ЫљЕУНсЙћЕУЕНСЫыФеѓСаЃЈ peptide arrayЃЉЪЕ бщШЗ ШЯ [50,51] ЁЃ зюНќ , Юв УЧНЋСПзгСІбЇ/ЗжзгСІбЇ ЃЈQM/MMЃЉдгЛЏМЦЫугУгкЬсИп OppA МА PSD95 ЕААз жЪгыЙбыФЧзКЯСІдЄВтЕФОЋЖШ ,врШЁЕУСЫГЩЙІ [15,16]ЁЃШЛЖј , етаЉбаОПЖМЪЧеыЖдЬиЖЈыФ/ЕААзжЪЬхЯЕПЊеЙЕФ ,ЫљЛёЕУЕФдЄВтФЃаЭНіЪЪгУгкЬиЖЈЮЪЬт ,ВЛОпЭЈгУадЁЃWoo КЭ Roux дјЗЂБэСЫвЛЬзбЯИёЕФыФ/ЕААзжЪНсКЯздгЩФмМЦЫуРэТл ,ЕЋвђашГЄГЬЖЏСІбЇФЃФтКЭИДдгШШСІбЇТЗОЖЗжНтЖјЯожЦСЫИУЗЈЕФЭЦЙу [52]ЁЃ
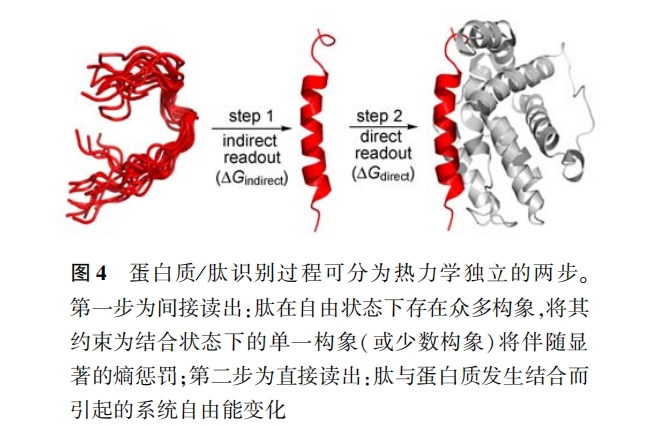
ДгддђЩЯРДЫЕ ,ОЋШЗдЄВтыФХфЛљНсКЯЕНЕААзжЪЪмЬхЙ§ГЬжаЫљАщЫцЕФздгЩФмБфЃЈ ЛђЧзКЯСІжЕЃЉашвЊЩюШыЦЪЮіИУЙ§ГЬЫљРњОЕФШШСІбЇТЗОЖЁЃ вЛАуПЩНЋЩњ ЮяЗжзгЪЖБ№Й§ГЬВ№ЗжЮЊСНИіШШСІбЇЖРСЂЕФВНжш :Ъз ЯШ ,ГЩдБЕїећЙЙЯѓвдДяЕНБэУцЛЅВЙ ,ЦфДЮ ,ЖўепЗЂЩњНсКЯЁЃ етСНВННдЖдЯЕЭГздгЩФмВњЩњЙБЯз ,ШЫУЧЯАЙпЩЯНЋЕквЛВНЙБЯзГЦЮЊМфНгЖСГіЃЈ indirect readoutЃЉ,Жј НЋЕкЖўВНЙБЯзНа зїжБНгЖСГіЃЈ direct readoutЃЉЁЃдч Цк,ЁА ОЕфЁБЩњЮяЗжзгзїгУРраЭШчЕААзжЪ/ЕААзжЪЁЂЕА АзжЪ/КЫЫсКЭЕААзжЪ/вЉЮяЕФжБНгКЭМфНгЖСГіЮЪЬтвбОЕУЕНСЫНЯЮЊЯЕЭГЕФВћЪіЁЃЬиБ№ЪЧШеБООХжнММЪѕбаОПЫљЕФ Sarai ПЮЬтзщПЊЗЂСЫгУгкЖЈСПМЦЫуЕААзжЪЖдDNA ЗжзгЪЖБ№Й§ГЬжажБНгКЭМфНгЖСГіФмЕФдкЯпЙЄОп ReadOut[53] ЁЃгЩгкЩЯЪіОЕфзїгУРраЭжаЕФМфНгЖСГіФмжївЊЪЧГЩдББфЙЙ Ыљв§Ц№ЕФгІБфФмЃЈ strain energyЃЉ,ЖјвЛАуШЯЮЊыФОпгаНЯДѓШсадЙЪВЛЛсдкНсКЯЙ§ГЬжаГіЯжУїЯдЕФгІБфФм ,СэЭтНсЙЙЗжЮівВЗЂЯжыФКмФбгеЕМЕААзЙЙЯѓИФБф [54] , вђДЫМфНгЖСГіЖдЕААз жЪ/ыФЪЖБ№ЙБЯзЩѕаЁЖјПЩвдКіТдЁЃ ШЛЖјетРяШДАЕВиаўЛњ :ОЁЙмгІБфФмЖдЕААзжЪ/ыФЪЖБ№ЙБЯзгаЯо ,ЕЋСэ вЛИіЗНУцМДвђШсадыФБЛЧЖШыЕНЕААзжЪЛюадПкДќФкЖјжТздгЩЖШЫ№ЪЇЫљв§Ц№ЕФьиГЭЗЃЃЈ entropic penaltyЃЉаЇгІШДВЛШнКіЪгЁЃ Шч Killian ЕШЩюШыЕїВщСЫШЫРр Tsg101 ЕААзгывЛИіОХыФЪЖБ№Й§ГЬКѓжИГі ,ьиГЭЗЃЖдИУЯЕЭГздгЩФмВЛРћгАЯьПЩИпДя 60 kcal.mol - 1 [55] ЁЃ МјгкДЫ ,ПЩвдШЯЮЊЕААзжЪ/ыФЪЖБ№ШдШЛДцдкВЛПЩКіЪг ЕФМфНгЖСГіЮЪЬт ,жЛВЛЙ§ДЫЪБьиГЭЗЃДњЬцСЫЭЈГЃЕФ гІБфФмГЭЗЃЃЈ ЭМ 4ЃЉЁЃФПЧА ,ЮвУЧе§дкЛљгкИУЙлЕуЗЂ еЙПьЫйЭЈгУЕФЕААзжЪ/ыФЧзКЯСІдЄВтЗНАИЁЃ
2. 3 ыФЕФЗжзгЖдНг
НЋИпЖШШсадЕФыФХфЛље§ШЗЖдНгШыЕААзжЪНсКЯПкДќЪЧвЛИіЬєеНЃЈ ЭМ 5ЃЉЁЃГЯШЛ ,ыФгыЕААзжЪОпгавЛжТЕФЛЏбЇзщГЩ ,гждкГпЖШЩЯНгНќвЉЮяЗжзг ,ЪЙЕУбаОПеп ЃЈ ЬиБ№ЪЧЪЕбщЙЄзїепЃЉШнвзвВЯАЙпгкНЋЯШЧАеыЖдЕААзжЪКЭвЉЮяЛЏКЯЮяЗЂеЙЦ№РДЕФГЩЪьЖдНгЗНЗЈжБНгЬзгУЕНыФРрЮяжЪЩЯЁЃШЛЖј ,ыФздЩэОоДѓШсадКЭЯпадЬи еїЪЙЕУетаЉЗНЗЈЭљЭљЕУВЛЕНРэЯыаЇЙћ ,ЩѕжСГіЯжЭъШЋЮѓЕМЕФНсЙћЁЃР§Шч ,гаШЫЯЕЭГЖдБШСЫЕБЧАСїааЕФ ЗжзгЖдНгЗНЗЈШч AutodockЁЂFlexxЁЂDOCK ЕШНЋыФХфЛљЁАдйЖдНгЃЈ redockЃЉЁБЕНЕААзЪмЬхЛюадПкДќжаЕФОЋЖШ ,НсЙћБэУїЫљгаЕФЗНЗЈаЇЙћЖМВЛОЁШчШЫвт ,ЬиБ№ЪЧИеадЖдНгЗНЗЈ DOcK МИКѕИјГіСЫЭъШЋ Дэ Юѓ ЕФ дЄ Вт [56] ЁЃСэвЛИібаОПвВЗЂЯж ,ЖдНгжЪСПЫцзХыФСДЕФдіГЄЖјбИЫйЖёЛЏ ,ЖрЪ§ЗНЗЈЮоЗЈе§ШЗДІРэДѓгк 3 ИіВа ЛљЕФыФХфЛљ [57] ЁЃетаЉбаОПжБЙлБэУїСЫыФЕФЙЬгаШсадМЋДѓЯожЦСЫГЃЙцЗНЗЈЖдЦфЕФЪЪгУад ЁЃСэЭт ,ШсадЕМжТЕААзжЪ/ыФНсКЯЙ§ГЬжаьиаЇгІЗЧГЃЯджј ,ЖјИУРр аЇгІЖдЯЕЭГздгЩФмЕФгАЯьдкДЋЭГЗНЗЈжавЊУДЪЧБЛЭъШЋКіТдСЫ ,вЊУДОЭЪЧЗЧГЃМђЛЏЕиДІРэЕєСЫ ЁЃЬиБ№ЪЧ , chang ЕШдк PNAS ЩЯзЋЮФжИГі ,ьиЖдЕААзжЪ/ХфЛљИДКЯЮяаЮГЩЕФгАЯьБЛДЋЭГЗНЗЈМЋДѓЕЭЙРСЫ [58] ЁЃЯдШЛ , ШчЙћХфЛљЪЧШсадыФ ,ФЧУДИУРрЮЪЬтНЋЛсИќЮЊбЯжи ,ЖјьигРдЖЪЧЯЕЭГШШСІбЇжазюФбвдзНУўЕФвђЫиЁЃ
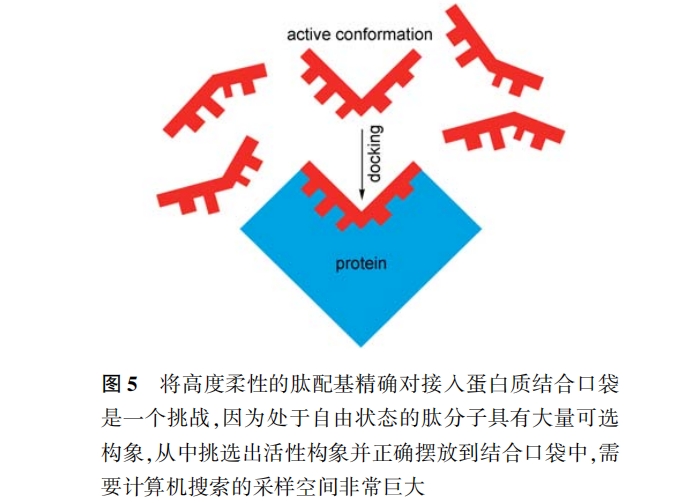
МјгквдЩЯдвђ , вбгаВПЗжбаОПепПЊЪМГЂЪдЗЂеЙзЈУХЕФыФЖдНгЗНЗЈ ЁЃдчЦкШЫУЧЬсГіВЩгУЦєЗЂЪНВпТдКЭжЧФмЫуЗЈЪЕЯжыФЖдНг [59] ,ЕЋШБЗІШЋУцВтЪдКЭЩюШыЦРМлЖјЮДЕУЕНЙуЗКгІгУ ЁЃКѓРДЖрЪ§баОПепМЏжагкВЩгУЗжзгЖЏСІбЇ/Monte carlo/ФЃФтЭЫЛ№ВЩбљгыНсЙЙгХЛЏЯрНсКЯЕФЗНЗЈДІРэЕААзжЪ/ыФНсКЯЮЪЬт ,ЬсГіСЫвЛЯЕСаыФЖдНгЗНЗЈ , Шч GІФ [60] ЁЂDocscheme [61] ЁЂ DynaDock [62] ЕШ , Ьи Б№ ЪЧвдЩЋСаЯЃВЎРДДѓбЇЕФ schueler-Furman МАЦфКЯзїепЪзДЮЪЕЯжСЫыФЖдНгЕФдкЯпЗўЮёЙЄОп FlexpepDock[63] ,ВтЪдБэУїИУЗЈЖдФГаЉ ЬхЯЕПЩвдДяЕНЁА бЧАЃЃЈ sub-angstromЃЉЁБМЖЕФЖдНгОЋЖШ[64] ЁЃзюНќ,Donsky ЕШвВЗЂВМСЫСэвЛИідкЯпыФЖдНгЙЄОп pepcrawler[65] ,ЮЊУцЯђгІгУФПЕФЬсЙЉСЫМЋДѓБуРћ ЁЃДЫЭт ,ыФЖЏЬЌЖдНгЗНАИВЛНігУгкдЄВтыФдкЕААзжЪЛюадПкДќжаЕФНсКЯЗНЪН ,ЛЙгУгкбаОПНсКЯЙ§ГЬЕФЖЏСІбЇЛњжЦЁЃAhmad ЕШВЩгУЖЏСІбЇЖдНгЪзДЮдкЗжзгЫЎЦНЩЯШЋГЬФЃФтСЫ sH3 гђгывЛИіЪЎыФЕФНсКЯЙ§ ГЬ ,ОнДЫЬсГіЕФЫЋЬЌФЃаЭАќРЈвЛИіГѕЦкПьЫйУжЩЂНзЖЮКЭвЛИіКѓЦкНчУцЫЎЗжзгХХИЩЙ§ГЬЃЛЫћУЧжИГіГЄГЬОВЕчаЇгІКЭЖЬГЬЪшЫЎСІЗжБ№ЪЧЭЦЖЏНсКЯЙ§ГЬЧАКѓСНИіНзЖЮЗЂеЙЕФЙиМќЧ§ЖЏвђЫи [66] ЁЃИУФЃаЭЕФећЬхТжРЊгыКѓРД staneva КЭ wallin РћгУШЋдзг Monte carlo ФЃФт pDZ гђЪЖБ№ЙбыФХфЛљНсТлЛљБОвЛжТ ,КѓепвВжЄЪЕСЫгђ/ыФНсКЯЙ§ГЬДцдкСНИіЗжБ№гЩОВЕчКЭЪшЫЎжЇХфЕФНзЖЮ [67] ЁЃЫљВЛЭЌЕФЪЧ Ahmad ЕШШЯЮЊЕквЛНзЖЮЗЧГЃЖЬднЧвШнвзгтдН ,Жј staneva ЕШШДШЯЮЊЕквЛНзЖЮЪЧЯоЫйВНжш ЁЃгаЪБЕБДѓСПЭЌРрЕААзжЪ/ыФИДКЯЮяНсЙЙвбжЊЕФЧщПіЯТЃЈШчЩЯЮФЬсЕНЕФ MHcЃЉ,ЖдНгЮЪЬтПЩвдМђЛЏЮЊЛљгкыФХфЛљЕФЙЋЙВжїСДЙЙЯѓдЄВтФПБъВрСД ,ШЛКѓдйзщКЯЦфЫћЗжзгФЃФтЗНЗЈЖдећЬхНсЙЙФЃаЭНјаааое§ , етбљПЩвд ДѓДѓЬсИпМЦЫуЕФаЇТЪКЭОЋЖШ [68]ЁЃ
Г§ДЫжЎЭт , ФПЧАеыЖдыФЖдНгЕФХфЬзбаОПЛЙЗЧГЃШБЗІ ,ШчЗЂеЙзЈУХЕФыФЖдНгЦРЗжКЏЪ§вдМАдЄВтыФЕФНсКЯЮЛЕуЕШ ЁЃжЕЕУвЛЬсЕФЪЧ,Petsalaki ЕШЛљгкЦНОљ ГЁТлЬсГіыФдкЕААзжЪБэУцЕФНсКЯЮЛЕудЄВтЗНЗЈЪЧИУ ЗНУцЕФживЊНјеЙ [69] ЁЃ
2. 4 ыФЕФЖЏСІбЇФЃФт
ЗжзгЖЏСІбЇФЃФтЃЈMDЃЉЪЧбаОПЩњЮяЗжзгЖЏЬЌааЮЊЕФОЕфЗНЗЈ , дјБЛЙуЗКгУгкЗжЮіыФЕФНсЙЙКЭаджЪЁЃбаОПзюЖрЕФЗНУцгІЪєЙбыФЕФелЕўЮЪЬт , вђЮЊМЦЫузЪдДЯожЦЖјЮоЗЈДгЭЗФЃФтЕААзжЪЕФЭъећелЕўЙ§ГЬ , вђДЫШЫУЧМФЯЃЭћгкбаОПЙбыФРДЮЊВћУїЕААзжЪелЕўааЮЊЬсЙЉгавцАяжњ [70] ЁЃШЛЖј,voelz ЕШЖд 872 ИіГЄЖШЮЊ8ЁЊ13 ЕФЕААзжЪЦЌЖЮФЃФтБэУїЙТСЂЙбыФелЕўЙ§ГЬНіФмВПЗжжиЯжИУыФЖЮдкЕААзжЪжаЕФПеМфНсЙЙаджЪ ,жЄУїСЫЕААзжЪелЕўЕФЗЧМгКЭад [71] ЁЃжЕЕУЧьавЕФЪЧ , ЫцзХМЦЫуЛњШэгВМўММЪѕЕФИпЫйЗЂеЙ ,ЯждкШЫУЧвбОФм ЙЛДгЭЗФЃФтаЁаЭЕААзЕФећИіелЕўЖЏСІбЇааЮЊЃЛШчзюНќ shaw ЕШдкбЧКСУыЪБМфГпЖШеЙЪОСЫ ww ыФЪЖБ№гђМАХЃвШЕААзУИвжжЦМС BPTI ЕФЭъећелЕўЙ§ГЬ[72] ЁЃДЫ Эт,MD вВБЛгУгкЗжЮіЕААзжЪ/ыФНсКЯШШСІбЇаджЪЃЈ ШчЙЙдььиБф[55] ЁЂНсКЯздгЩФм [73] ЕШЃЉвдМАыФгыЦфЫћЮяжЪЃЈ ШчБэУцЛюадМС[74] ЁЂФЩУзВФСЯ [75] ЕШЃЉЕФзїгУааЮЊ ЁЃетРяашвЊЬсЕНСНИігаШЄЕФЙЄзї:ЃЈ1ЃЉLama ЕШЭЈЙ§ФЃФтЖдБШРДздгкПЙЕђЭіЕААз BH3 ЕФВЛЭЌТна§ыФдкздгЩзДЬЌЯТвдМАдкгы B ЯИАћСмАЭСівђзг 2ЃЈ Bcl- 2ЃЉНсКЯзДЬЌЯТЕФЖЏСІбЇаджЪ ,ЗЂЯжЖд Bcl-2 ЧзКЯСІ НЯИпЕФСНИіыФФмЙЛдкЮДНсКЯзДЬЌЯТВПЗжЮЌГжЦфТна§ЙЙЯѓ , вдДЫМѕ ЩйНсКЯЕМжТЕФЙЙЯѓьиЫ№ЪЇ [76] ЁЃЃЈ 2 ЃЉ Dagliyan ЕШВЩгУРыЩЂЗжзгЖЏСІбЇЃЈ discrete molecular dynamicsЃЉМгЫйФЃФтСЫЪ§ИіыФЗжзггыЦфЬьШЛЪмЬхЕФ НсКЯЙ§ГЬ , НсЙћВЛЕЋжиЯжСЫЩЯУцЬсЕНЕФЫЋЬЌФЃаЭ ЃЈ ВЮМћ 2. 3 НкЃЉ,ЖјЧвдкдзгЫЎЦНЩЯОЋЯИЙДРеСЫИУЙ§ГЬжаыФВрСДЙЙЯѓКЭЕААзжЪНсКЯЮЛЕуНсЙЙЕФБфЛЏ ЧщПі [77] ЁЃ
ДЫЭт ,ыФЕФЖЏСІбЇФЃФтвВГЃБЛгУгкыФЖдНгвтЭМ , ЛђгУгкОЋаоыФЖдНгЫљЛёЕУЕФГѕЪМНсЙЙЃЛЫфШЛДЫЗЈаЇТЪНЯЕЭ ,ЕЋЪЧЭљЭљФмЙЛЕУЕННЯЮЊПЩППЕФЕААзжЪ/ыФИД КЯЮяФЃаЭ , вђДЫврГЃВЩгУ [62] ЁЃ
2. 5 ыФЕФНсЙЙЪ§ОнЗжЮі
ЫцзХ PDB Ъ§ОнПт[37] жаОЇЬхбЇЛђЖрЮЌ NMR ММЪѕНтЮіЕФыФгыЕААзжЪЫљГЩИДКЯЮяНсЙЙПьЫйдіГЄ ,ДгетаЉЪЕбщНсЙЙЪ§ОнжаЬсШЁКЭЙщФЩгагУжЊЪЖЮЊНтЪЭЕА АзжЪ/ыФЪЖБ№КЭЯрЛЅзїгУааЮЊМАдЄВтЧБдкЕФзїгУФЃЪНЬсЙЉСЫЗЧГЃгаМлжЕЕФВЮПМаХЯЂ [78] ЁЃvanhee ЕШЕї ВщСЫ 731 ИівбжЊНсЙЙЕФЕААзжЪ/ыФНчУцКѓШЯЮЊыФгы ЕААзжЪ НсКЯЗНЪНЗЧГЃРрЫЦгкЕЅЬхЕААзЕФелЕўФЃЪН[79] ,ИУЗЂЯжЮЊЛљгкЗсИЛЕААзНсЙЙЪ§ОнЩшМЦЕААз жЪ/ыФЯр ЛЅзїгУЬсЙЉСЫРэТлвРОн ЁЃШЛЖј , Щд Кѓ London ЕШНјвЛВНЩюШыЗжЮівЛзщИпжЪСПбљБОКѓжИГі , ыФЭљЭљВЩгУБШЕААзелЕўИќРЮЙЬЕФЗНЪНгыЪмЬхНсКЯ , ДгЖјУжВЙАщЫцИУЙ§ГЬПЩЙлЕФьиГЭЗЃ [54] ЃЛДЫЭтЫћУЧЛЙЗЂЯжвЛаЉгаШЄЕФЯжЯѓ , БШШчыФХфЛљЭЈГЃНсКЯЕНЕААзжЪБэУцзюДѓАМЯнДІ ,ЕААзжЪ/ыФНчУцГЃДцдкЙиМќадЕФ ЁА ШШЕуВаЛљЃЈ hotspot residueЃЉЁБЕШ ЁЃСэЭтвЛаЉбаОПепЭЈЙ§ПМВьЕААзжЪИДКЯЮяНсЙЙРДбаОПыФЕїНкЕФЕААзжЪЯрЛЅзїгУ ,Шч Jochim КЭ Arora ЭЈЙ§ЖдНќЭђИіЖрдЊЕААзИДКЯЮяМьЪгЗЂЯжЦфжадМга 13% ЕФНчУцАќКЌТна§ФЃЬх ,АЕЪОСЫТна§ыФЪЧЗЧГЃгаЯЃЭћЕФЕААзИДКЯЬхзА ХфЕФвжжЦЙЙМм [80] ЁЃЕФШЗ ,ЦљНёЪЕбщШЗШЯЕФАаБъЛюадыФЖрЪ§ЮЊТна§ад ,ЭЦВтетПЩФмЪЧвђИУРрЖўМЖНсЙЙЮШЖЈЛЏГЬЖШНЯИпЧввзгкЛЏбЇдМЪјжЎЙЪ [81] ЁЃДЫКѓвЛаЉбаОПЗЂЯж ,Г§СЫТна§ФЃЬхжЎЭт ,ЦфЫћРраЭЕФЯпадађСа ЃЈ ШчЛЗзДађСаЃЉвВГЃГіЯжгкЕААзжЪИДКЯЮяНчУцВЂЖдКѓепаЮГЩЦ№ЕНСЫживЊЕФЭЦЖЏаЇгІЃЛгЩДЫЙРМЦыФЖЮЕїНкЕФЕААзжЪЯрЛЅзїгУПЩЖрДя 50% ,ЧвЫќУЧЖМЪЧЧБдкЕФЕААзжЪИДКЯЮязАХфЁА здвжжЦыФЃЈ self-inhibitory peptideЃЉЁБ[82] ЁЃШЛЖј ,ЩЯЪіНсТлашвЊПМТЧИќЖрЕФвђЫиВХгавтвх ,Шч stein ЕШЭЈЙ§ЖдДѓСПыФЕїНкЕФЕААзжЪЯрЛЅзїгУНјааЭиЦЫФмСПЗжЮіКѓжИГіКЫаФЯпадађСаЭљЭљашвЊдкЬиЖЈЕФНчУцЛЗОГжаВХФмБЃжЄИпЕФЬивьадКЭЪЪЕБЕФЮШЖЈад ,ЛЗОГвђЫиЦНОљЙБЯзСЫ 20% вдЩЯЕФЧзКЯ СІ[83] ЁЃвђДЫЩшМЦздвжжЦыФЪБШчКЮАќКЌНчУцЛЗОГЙБ ЯзЪЧЩшМЦепашвЊПМТЧЕФжївЊвђЫижЎвЛЁЃ
2. 6 ыФЕФЗжзгЩшМЦКЭНсЙЙаоЪЮ
РэадЩшМЦЬиЖЈыФЗжзгВЂЖдЦфНсЙЙНјаааоЪЮИФдь ,ЪЙжЎЛёЕУИпЕФАаБъЧзКЯСІКЭбЁдёадвдМАСМКУЕФДњаЛЮШЖЈадКЭФЄДЉЭИФмСІЪЧыФРрвЉЮябаЗЂепзЗЧѓЕФУЮЯы ЁЃЗНЗЈбЇЗНУц , ШЫУЧЬсГіСЫRosetta [84] КЭ vitAL [85] ЕШЗНАИНјааШЋаТЃЈ de novoЃЉыФХфЛљЩшМЦ ,Жј ЮвУЧвВдјПЊЗЂСЫ LigEvolutioner ГЬађгУгквбжЊЯШЕМыФЃЈ lead peptideЃЉЕФНсЙЙздЖЏНјЛЏИФСМ[86] ЁЃНќФъРД , РэадыФЩшМЦвргыИїРрЩњЮяЛюадМьВтЪЕбщНєУмНсКЯЦ№РД ,етаЉЙЄзїГЩЙІЕиНЋРэТлЭЦЯђСЫгІгУ ЁЃЫќУЧжївЊ дкШ§ИіВуДЮЩЯЕУЕНЪЕЯж :вЛЪЧЛљгкађСажЊЪЖ ,ЭЈЙ§ЭГМЦбЇЯАФЃаЭКЭЩњЮяаХЯЂбЇЗНЗЈЬсСЖКЭгХЛЏФПБъыФађСа ,Шч Edwards ЕШЩИбЁбЊаЁ АхЛюадЕїНквђзг[87] , shemesh ЕШЗЂЯжGЕААзХМСЊЪмЬхМЄЖЏМС [88]вдМА walshe ЕШШЗЖЈШЫРрАзЯИАћПЙдНсКЯБэЮЛ[89] ЕШЪЧИУЫЎЦНбаОПЕФЕфаЭДњБэЃЛЖўЪЧЛљгкФЃК§НсЙЙаХЯЂ ,ЭЈЙ§ађСаЭЦВтАаБъЕААзЕФГЃМћНсЙЙФЃМм ,дђПЩИљОнвбжЊЕФДѓСПИУРрФЃМмНсЙЙЖЈвхФПБъыФ ,Шч kliger ЕШВЩгУ Fourier БфЛЛЩшМЦАщТТЕААзЕФЛюадЕїНкыФ[90] МА yinЕШПЊЗЂЕФећКЯЫиПчФЄЧјАаЯђыФ [91] ЁЃКѓепПЩЪгЮЊИУСьгђЕФЭЛЦЦадЙЄзї , вђЮЊбаОПепдкЭъШЋЮДжЊећКЯЫиПчФЄНсЙЙЕФЧщПіЯТНіРћгУвбжЊЩйЪ§ФЄЕААзЕФПчФЄТна§ПеМфХХСаФЃЪНОЭЩшМЦГіСЫФмЙЛздЖЏзАХфЕНЯИАћФЄФкВЂгыећКЯЫиАаБъЧјгђЗЂЩњЬивьадзїгУЕФЛюадыФЃЛШ§ЪЧЛљгкОЋШЗАаБъНсЙЙЪ§Он ,ЭЈЙ§ЗжзгФЃФтЗНАИКЭФмСПЗжЮіВпТдЛёЕУИпадФмЕФыФХфЛљ ,Шч cui ЕШЗЂЯжДйжзСіЕђЭіыФ [92] ,sood ЙЙдьжзСіЕААзЕїНкыФ[93] вдМА Grigoryan ЕШЩшМЦССАБЫсРСДЃЈbZIPЃЉЬивьЙЅЛїыФ [94] ЁЃКѓвЛИіЙЄзїЬиБ№гаШЄ , вђЮЊзїепЪзДЮБЈЕРСЫЖЈЯђЩшМЦыФЕФЬивьадЃЈЖјЗЧЧзКЯСІЃЉЕФГЩЙІАИР§ЁЃ
2. 7 ыФЕФЯЕЭГЩњЮябЇааЮЊбаОП
ЯрЖдЩЯЪіЬиЖЈАИР§ЗжЮіЖјбд ,ЯЕЭГЩњЮябЇбаОПыФЕФаджЪКЭЛюадИќзХблгкдкећИіЛљвђзщЫЎЦНЁЂећИіЯИАћЭјТчКЭећИіМвзхЗжРржаПМВьыФгыАаБъжЎМфЕФИДдгзїгУааЮЊКЭЖрЮЌЯрЛЅЙиЯЕ [95] ЁЃвЛАуВЩгУИпЭЈСПЪЕбщЪжЖЮ , ШчЪЩОњЬхеЙЪОыФПт[96] КЭ sPOT КЯГЩММЪѕ [97] ,ПЩвддкЖЬЪБМфФкВњЩњДѓСПЕФКђбЁыФ ,НјЖјЩИбЁЫќУЧЖдЬиЖЈАаБъЃЈ ЛђМвзхАаБъЃЉЕФЧзКЯЧБСІ[98] ЁЃетаЉЪ§ОнЪЧживЊЕФЯЕЭГыФЩњЮябЇбаОПзЪдД ЁЃЖјЛљгкађСаКЭНсЙЙВњЩњЕФРэТлФЃаЭврБЛгУгкЙЙдьЬиЖЈАаБъЕФЭъећЛљвђзщЪЖБ№ЦЪУц [99,100] ЁЃДЫЭт ,ДгађСаЁЂНсЙЙКЭзїгУЖдЯѓНЧЖШЬНЬжФГвЛРрыФЪЖБ№гђЕФМвзхФкВПЙиСЊадКЭИіЬхМфВювьадврЪЧдкЯЕЭГЫЎЦНбаОПгђ/ыФЗжзгНјЛЏЕФГЃМћВпТд [101,102] ЁЃЖдДЫ , етРяНіЬсМА stiffler ЕШЕФЙЄзї[103] :ЫћУЧВЩгУЧзКЯСІМьВтКЭжїГЩЗжОлРрЯрНсКЯЕФАьЗЈбаОПСЫаЁЪѓЬхФкБэДяЕФ 157 жжPDZ гђдкХфЛљбЁдёадПеМфЕФЗжВМЧщПі ,НсЙћБэУї ,ећИіPDZ гђМвзхЭЈЙ§НјЛЏгХЛЏКѓГЪОљдШЗжЩЂдкИУПеМфжа ,ДгЖјзюДѓЯоЖШБмУтСЫИіЬхжЎМфЕФНЛВцЗДгІ ЃЈ cross-reactivity ЃЉЁЃ НќФъРДаЫЦ№ЕФыФ зщбЇЃЈpeptidomicsЃЉвдМАдкДЫЛљДЁЩЯЗЂеЙЦ№РДЕФыФзщбЇПтКЭЪ§ОнЭкОђММЪѕПЩЮНКѓЦ№жЎау ,БиНЋдкЮДРДДѓЗХвьВЪ [104,105] ЁЃ
3 змНсМАеЙЭћ
ЫцзХИпЭЈСПВтађММЪѕКЭЯЕЭГзщбЇЗНЗЈЕФИпЫйЗЂеЙ,kahvejian ЕШдкЦфЧАеАадТлжјжадЄВт ,ВЛГі 20 ФъШЫУЧНЋВтЖЈМИКѕЫљгаЕФЩњЮябЇЪ§Он ЁЊ жСЩйПЩвдЫцвтВтЖЈздМКЯывЊЕФЪ§Он [106] ЁЃДгЖј , ЖрЪ§ЩњЮябЇМвЖМЃЈЛђЖрЛђЩйЃЉж№НЅзЊБфГЩЮЊМЦЫуЩњЮябЇМв , вђЮЊФЧЪБЩњЮябЇМвЕФжївЊОЋСІЛсгУгкРћгУМЦЫуЙЄОпДгКЃСПЪ§ОнжаЗЂОђЩњЮябЇжЊЪЖ ЁЊ е§Шч Jorgensen аћГЦЕФФЧбљ [107] :we, re all computational biologists! ОЁЙметбљЕФТлЖЯЙ§гкМЄНј ,ЕЋЯђШЫУЧебЪОСЫРэТлКЭМЦЫуЗНЗЈвдМАаХЯЂЭкОђММЪѕдкЩњУќМАЦфЯрЙибЇПЦжаЕФЙуРЋгІгУЧАОА ЁЃНќФъРД , гЩгкыФдкЯИАћаХКХзЊЕМжаЫљАчбнЕФживЊНЧЩЋвдМАзїЮЊЩњЮявЉЮяАаЯђЕААзжЪЯрЛЅзїгУЭјТчЕШЬиЪтаджЪЕФЗЂЯж ,ЛНЦ№СЫПЦбЇЙВЭЌЬхЖдыФЕФХЈКёаЫШЄ ЁЃгыжЎЯрАщЕФЪЧ ,МЦЫуЛЏбЇКЭЩњЮяаХЯЂбЇСьгђгыыФЯрЙиЕФбаОПЙЄзїШевцдіЖр ,ВЂШЁЕУСЫГЄзуНјеЙ ЁЃМјгкыФЕФРэТлМЦЫубаОППЊЪМЦ№ВН ,ВЂвбНЅНјМбОГ ,БОЮФвдМЦЫуыФбЇЮЊжїЬтИХРЈИУСьгђЕФбаОПЗЖГыКЭбаОПЖЏЬЌ ,ЦфФПЕФЪЧЯЃЭћНЋжЎЯЕЭГЛЏЮЊвЛИізЈУХЗжжЇбЇПЦБугкЯрЙибаОПепНЛСїЬНЬж ЁЃПЩвддЄ Мћ ,МЦЫуыФбЇЕФЗЂеЙБиНЋЪмЕНМЦЫуПЦбЇКЭыФПЦбЇСНЗНУцНјеЙЕФЭЦЖЏ ,НёКѓЦфбаОПжиЕугІИУжївЊМЏжадкЯТСаМИИіЗНУц:ЃЈ1ЃЉЫцзХЕААзжЪ/ыФИДКЯЮяШ§ЮЌНсЙЙЪ§ОнЕФШевцРлЛ§ ,дкдзгЫЎЦНПМВьыФгыЕААзжЪЪЖБ№КЭЯрЛЅзїгУЕФРэЛЏЛљДЁМАШШСІбЇаджЪНЋЕУЕННјвЛВНЗЂеЙЁЃЃЈ2ЃЉВЩгУЗжзгЩшМЦЗНЗЈЛёЕУИпадФмыФФЃФтЮяЙЅЛїЕААзжЪЯрЛЅзїгУЭјТчвбПЊЪМеИТЖЭЗНЧ ,ЮДРДгаЭћГЩЮЊаТвЉбаЗЂЕФвЛИіИпЫйдіГЄЕуЁЃЃЈ3ЃЉЙЙНЈыФзщбЇЪ§ОнПтМАЭкОђФкКаХЯЂПЩдкЯЕЭГЩњЮябЇВуДЮВћЪЭЯИАћаХКХзЊЕММАДњаЛЭЈТЗжаИїРрыФЕФЙІФмгыЛюадЁЃЃЈ4ЃЉвдыФЮЊЛљДЁПЊЗЂздзщзАФЩУзЙмЁЂБэУцЛюадВФСЯЁЂ вЉЮяДЋЪфЯЕЭГЕШЯрЙиЪЕбщбаОПвбгаБЈЕР ,ЯраХМЦЫу ФЃФтКЭРэТлЩшМЦЙЄзїНЋдкВЛОУжЎКѓЙуЗКеЙПЊЁЃ
Утд№ЩљУїЃКБОЮФЮЊаавЕНЛСїбЇЯАЃЌАцШЈЙщдзїепМАддгжОЫљгаЃЌШчгаЧжШЈЃЌПЩСЊЯЕЩОГ§ЁЃЮФеТБъзЂгазїепМАЮФеТГіДІЃЌШчашдФЖСдЮФМАВЮПМЮФЯзЃЌПЩдФЖСддгжОЁЃ
