
ժҪ�����滯���������Բ������Ӧ���������ߵ�����Ч�����ޡ�ǰ��ҩ����һ��ͨ�������ӽ�ҩ���������� (�翹�塢���ġ��������塢�ۺ����)���ӳɵ�ҩ��ż��������ҩ����������λ���͵�Ч�ʣ������ҩ�����Ч�Ͱ�ȫ�ԡ����Ľ����˼��ֿ����������������Ƶ�ǰ��ҩ��翹��-ҩ��ż�������-ҩ��ż�����������-ҩ��ż����;ۺ���-ҩ��ż��������������ɡ������ҩԭ�����ٴ��о���չ�����в�Ʒ����������ǰҩ�������ٴ�Ӧ���д��ڵ����⣬����Ϊǰ��ҩ���з��ṩ�ο���
����������һ�����Լ�����Ҳ����в�������������ij����������ݱ�����2018 �꣬ȫ������������·��������ӵ� 1 810 �����������������ӵ�960 ���� [1]���ڶ��������������У������dz������������Ҫ���Ʒ�����ʹ��С���ӿ�����ҩ����Ч��ֹ���������������������Ȼ�����ܴ�һ����С���ӿ�����ҩ�����߱�ʶ������ϸ������������ͬʱ����������ϸ�����Ӷ����½ϴ�IJ�����Ӧ [2]�������ִ���ҽ�Ƶ�������������ҩ��Ŀ����������о��ߵĹ㷺��ע����һ�����ǽ���С���ӿ�����ҩ������Ӧ�����������Ч����Ч������������ʮ��ķ�չ�����˽�������ҩϵͳ ( �罺����֬���塢�������� ) ʵ��ҩ��İ�������⣬ǰ��ҩ��Ҳ��ʵ�����������ҩ��һ����Ҫ���� [3]��ǰ��ҩ���ǽ���������Ե�ԭ��ҩ��ij�ַ���Ƭ���ϵĻ�ѧ����ͨ�����ۼ���ϵõ���ż��������γɵ��»�ѧʵ�屾�����Ի���Ե���ԭ��ҩ���������ھ���ø���û�ѧ��Ӧ��ʹ������ϼ��ѽ⣬�ͳ�ԭ��ҩ�������������� [4]�����������о����ֽ�����������λ�����Ա���İб��������ϵķ�����ҩ��ͨ�������������γ�С������������ǰ��ҩ��������������ҩ����С���Ӱ���ҩ����ȣ���ԭ�����Ұ���Ч�ʸߡ�������ҩ ����������ҩϵͳ��ȣ����Ʊ���������ԭ�ԣ��ɴ�����������������ٴ�ת����Ŀǰ�����������������Ƶ�ǰ��ҩ����Ҫ�������� - ҩ��ż���� (antibody-drug conjugate��ADC)������ - ҩ��ż���� (peptide-drug conjugate��PDC)���������� - ҩ��ż���� (aptamer-drug conjugate��ApDC) �;ۺ��� - ҩ��ż���� (polymer-drug conjugate) �ȡ�
1 ���� - ҩ��ż���� (ADC)
���� 20 ���ͳ����ԡ�����֮������������ �� ����ϣ (Paul EHRLICH) Ϊ�����Ŀ�ѧ���Ǿ�����ˡ�ħ���ӵ������ۣ�ּ��Ѱ��һ�ֻ�������ѡ�������λ������������������Ӧ [5]��20 ����70 ��������ڵ���¡���� (monoclonal antibodies��mAbs) ���������ƿ�ʼ���� [6]��mAbs �ɼ��ٷ������Զ��ԣ�ͨ�������Խ������ϸ���ϵĿ�ԭ���������ض��ź�ͨ·�Դﵽ����Ч������ֱ�Ӷ�����ϸ���������߷�Ӧ [7]������Ϊֹ��Լ�� 30 �� mAbs�������FDA�������������ơ�Ϊ�������Ч�����о����ֽ� mAbs ����ֿ�����ЧӦ���� ( ��ϸ����ҩ������Ժ��ء����߶��ص� ) �������ӣ���������� mAbs �İ������ƺ��������ƣ����� ADC�ܵ��㷺��ע [7]��
ADC ������ mAbs ͨ����������ϸ����ҩ�ﹲ�۽���γɣ����� mAbs �ɽ�ҩ�������͵�Ŀ��ϸ���з������� [ 8]�����ֽ����ϸ����С����ҩ�������ż���� [ ��Է�������Ϊ 300 �� 1 000��������Ħ�����İ�������Ũ�� (IC50) ] �߱���Ч��������ϸ��������������ͬʱ mAbs �ָ������ѡ���ԡ��ȶ��Ժ�����ҩ��ѧ�����ԣ�������Ҫ����Ϊ��Ѫѭ��ʱ�䳤���������Ը߶����е�����Ч��ǿ�Ͷ�������֯���Ե͡�������ҩ������ԭ������ [9]��
1.1 ���û���
ADC �Ŀڷ��������öȽϲ��Ҫ����ע���Ա���θ��������ø�Կ���Ľ��⡣ADC ��Ѫ�����е� mAbs �ɷֿ����ϸ���ϸ߱���ı��濹ԭʶ��ϣ�ͨ�����鵼�����������ڻ����γɺ��� ADC- �п�ԭ����������ں����塣�����ں������У����������� (ATP) ���������ӱò������Ի���������һ���� ADC �� mAbs �� Fc Ƭ����������б���������� Fc ���� (FcRn) ��ϣ�������ϸ��Ĥ�ں϶����ų�ϸ���⡣������ں���������ø���ںϣ�δ���ų������ ADC ����ø��ˮ��ø�������£��ͳ�ϸ����ҩ�ﵽϸ�����С�ϸ����ҩ���� DNA���ܵ��Ƚ�Ϻ���ϸ�� DNA���ƻ���˿�������裬��������ϸ��������Ŀ��ϸ������ʱ���ͷŵ�ϸ����ҩ�ﻹ��ͨ���Թ���ЧӦ (bystander effect) ������Χ����ϸ������Χ������֯����������ЧӦȡ����ϸ����ҩ�����ˮ���� [10]��ADC �鵼��ЧӦ�����������ϵͳ��ͨ�����ֻ��ƴ�������ЧӦϸ������������λ�� [11]��
1.2 �������
1.2.1 mAbs
mAbs �� 2 ���鵼��ԭʶ��Ŀ�ԭ���Ƭ�� ( Ҳ����Ϊ Fabs) �� 1 ���鵼����������ϵͳЧӦϸ������õĺ㶨Ƭ�� ( �� Fc Ƭ�� )��Fc Ƭ���а����� FcRn ��ϵĽ�������Ե��ڿ�����ѪҺ�еİ�˥�� [12]��ADC �е� mAbs ͨ����߱��������� [13] ���ٽ�С������ԭ�ԣ�ͨ��ѡ����Դ����ȫ��Դ���Ŀ��� ���ڰ��������� ( �����㹻�Ŀ�ԭ�����Ժ����� ) ��Ч��ϸ���ڻ����� ����ѭ����˥�ڳ���
ADC ��������Ҫ������Ҫȷ������֤ mAbs ����Ӧ�Ŀ�ԭ�б꣬�ڿ�ԭѡ������Ҫ�������¼������� [14]�����ȣ�Ŀ�꿹ԭӦ������ϸ�����棬�Ա�ADC-�п�ԭ��������˳���ڻ�����ϸ������ҩ����Σ������Ŀ�꿹ԭӦ��Ŀ��ϸ��������ȱ�����ڽ�����֯�б������ϵ� �����Ŀ�꿹ԭ������Ӧ�����٣��Է�ֹ���뿹ԭ��ѪҺѭ������mAbs ��ϣ����� ADC ʧЧ��Ŀǰ��������ٴ��о��� ADC �б�ܶ࣬��Ҫ��ΪѪҺ�����б��ʵ�����бꡣ���磬�������ڰ�Ѫ���İб���CD22��CD30��CD33 �ȣ�����ʵ���������˱�Ƥ������������ -2(HER2)�����ӵ��� -4(nectin-4)��ǰ����������Ĥ��ԭ (PSMA)����Ƥ������������(EGFR) �� [15]��
1.2.2 ϸ����ҩ��
ϸ����ҩ���� ADC ������ЧӦ�ɷ֣������Ʊ� ADC ��ҩ��ͨ����߱��������� [16] �������û�����ȷ ���ھ��м�ǿ��ϸ������ ( IC50 ����Ħ�������� ) ���ۿɱ�ֱ�����Σ���ṹ�������п����ӻ�����������Բ���Ӱ�� ������ mAbs ��Һ�����ȶ����ڲ�����ܽ⡣
Ŀǰ�ٴ�Ӧ������ϸ����ҩ����������û��ƿɷ�Ϊ������ [17]���� DNA ���˼� ������ù����(calicheamicins��CLM)��������� (doxorubicin��DOX)������ù�� (duocarmycins)��������������䓬�� (pyrrolobenzodiazepines��PBDs) �ȣ���Щҩ��ͨ���� DNA ˫����С����ϣ����� DNA �ѽ��ϸ�����������ܵ������Ƽ����������� (maytansines)�Ͱ�����͡ (auristatins) �ȣ�ͨ�����ܽ�϶���ֹ�ܵľۺϡ�����ϸ�����ڣ��̶��յ�ϸ���������������칹ø���Ƽ� ��ϲ���� (camptothecin��CPT) ���������ͨ������ DNA ����������������칹ø��ʹ DNA �����ѡ�
1.2.3 ������
��ѧ�����ӵ������ǽ�ϸ����ҩ���� mAbs ���ӣ�������ѭ����ά�� ADC ���ȶ��ԡ������ӵĻ�ѧ���ʺ�ż��λ����� ADC ���ȶ��ԡ�ҩ��ѧ��ҩЧѧ���Լ����ƴ��ȷ������������Ҫ�����á������ӵ�����Ӧ���� [18] �����㹻���ȶ��ԣ�ʹ ADC ����Ѫ����ѭ������λ���в�λ�����������Ѷ����·������Զ��� ���������ڻ�������Ѹ�ٶ��ѣ��ͷ�ϸ����ҩ�
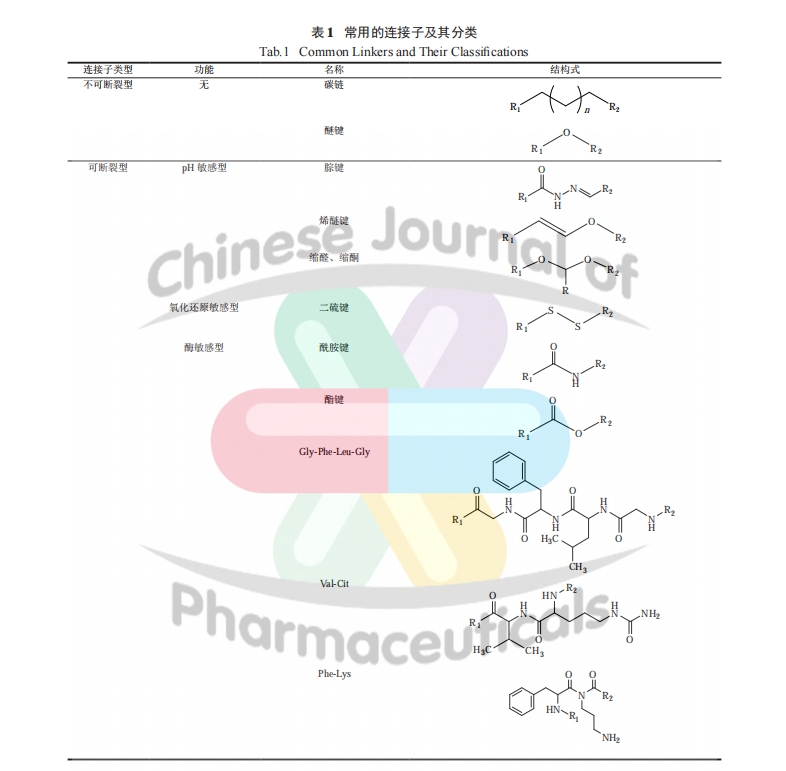
����ҩ���ͷŻ��ƣ�Ŀǰ���õ������ӷ�Ϊ�ɶ����ͺͲ��ɶ����� ( �� 1) [19]���ɶ����͵������������Ի��� [ ������� (pH 5.0 �� 6.0) ����ø�� ( pH 4.0 �� 5.0) ] �в��ȶ���ͨ�������л���( ����� ) ��ˮ�⡢����øˮ�� [ ��������ϸ���ڸ߶ȱ������ø�嵰��ø B ʶ���ѽ��ض����ļ������Ӱ��� - �ϰ��� (Val-Cit��VC) ���� ] ��ԭ��Ӧ [ ����ϸ���ڵĸ�Ũ�ȹ����� (GSH) ��ԭ�������еĶ������ʹ����� ] �����ã���ҩ��� ADC ����Ч�ͷų��������ɶ�������������ͨ�� mAbs �ϵİ�����л���ҩ���γɲ��ɻ�ԭ�Ļ�ѧ���������ѪҺ�и��ȶ������ɶ����������� [ �������鵥�� - ��̹��ż���� T-DM1(ado-trastuzumab emtansine) �е����������� ] �����ڵ����������ø�彵�����ͷ�ҩ������Ҫ�����Ч���ڻ������ŵ���ø��ת��·����
1.3 �ٴ�����
ADC ������� 1958 �꣬����ʱ�������ֱ�� 1983 ��ų��� ADC ���״��ٴ����鱨��������Դ�����߿�ԭ (CEA) �����볤���¼�ż�����������ڶ������������� [8]�����������ϵ� ADC �ǻ��Ϲ�˾�з��ļ����鵥���������� (gemtuzumab ozogamicin����Ʒ�� Mylotarg)������ 2000 ������� FDA �����У�����������Դ���� CD33 ��������ϸ�����ؿ���ù��ż�����ɣ��������Ƽ�����ϸ����Ѫ�����������ٴ�Ӧ���о�������������Ӧ�Ļ�ѧҩ��ȣ�����������ߵ������ʣ��������ز�����Ӧ������� 2010 �곷�� [20]��Mylotarg ��ʧ��֮�����ڣ������õ�����������Ļ�ѧ���ʲ��ȶ�����δ�ﵽ�е�ʱ�ͱ�ˮ�⡣�����������Ӳ��ȶ��IJ���ԭ�������������ż��λ�ò�ȷ��������ʵ�ֶ���ż�����������ҩЧ���ޡ����ܵ�һ��ADC ��ʧ�ܸ��գ�����Ϊ���� ADC �ķ�չ�춨�˻�����
�� Mylotarg ֮��ά���������� (brentuximab vedotin����Ʒ�� Adcetris) �� 2011 ������������ƻ�����ܰ�����ϵͳ�Լ���Դ�ϸ���ܰ��� [21]��T-DM1( ��Ʒ�� Kadcyla) �� 2013 ����������FDA �������������� HER2 ���Ե�ת�������ٰ� [22]������Ϊֹ������ FDA �Ѿ��������� 7 ��ADC( ���� 2)������ 100 ��� ADC ҩ�����ڿ�չ�ٴ��о� [23��26]��

���� ADC ҩ����з���ȡ�ýϴ�ͻ�ƣ����Դ����������� ���ٿ������ҩ�� (drug-to-antibody ratio��DAR) ���⣬Ŀǰ�õ��ľ��Ǻ��в�ͬ DARֵ ( 0 �� 8) �Ķ��ֳɷֻ���һ�� DAR Ϊ 3 ��4 �Ϻ��ʣ�DAR>4 ����ֽϵ͵������ԡ��ϸߵ�Ѫ������ʺͽϵ͵����ڹ�Ч ����Ŀǰ�õ���ͨ����ż��λ������Ŀ�����Ļ���δ����ȫʵ�ֶ���ż����ҩ�������ȷ��������ҩ��ѧ�о��������� ���ۿɶ�������������ѪҺ����ǰ�Ͽ�����ϸ����ҩ��û��ѡ���ԣ��ʴ������صĸζ��ԵȲ�����Ӧ���������ƴ���խ���� ADC ����֯���������ޣ�������ʵ���������д���һ�����ѣ�Ҫ���ǿ����С�ͻ�����ȡ�
2 ���� - ҩ��ż���� (PDC)
PDC �����ԭ���� ADC ���ƣ���Ҫ����ҩ����ͺ���������֮ͬ������ ADC �еĿ���ɷֱ�����Ϊ��������Ķ��ķ�����ȡ������ ADC��ȣ�PDC ����������� ��ͨ��������ҩ���Ƽ������Ʊ����ɱ��ϵ��Ҵ����������ԭ�ԣ����������������߷�Ӧ [27]��PDC �ĺϳɡ�������ʿ�������ף��������ȶ��Ը��ã����� PDC ��Է�������С���и��õ�Ѫ�ܡ���֯��ϸ����ͨ�ԣ������������ [28]��ֵ��ע����ǣ�������С����˹����ã�PDC ��Ѫ��������ٶȿ��� ADC�������� PDC ;���в�λ��ѭ��Ƶ�Σ����ܻ�Ӱ�������Ч�� [29]��
2.1 �������
2.1.1 ����
���������ŵ�������ѧ���ɾ���չʾ�Ͷ��Ĺ���ϳɵȼ����Ŀ��ٷ�չ��Խ��Խ������Ͷ��ı����ֻ�����ƣ�����شٽ��� PDC �ķ�չ��PDC �еİ�����ķ�����Ҫ���Ӧ�б������Ħ������������ (��ϳ���С�� 10�C9 mol/L)[30]���Ӷ�������õ�ѡ���ԣ��Լ���ȫ����ҩʱ��ɵķ������Էֲ��붾�� [31]��
���� PDC �Ķ��ķ���һ��ɷ�Ϊϸ������ (cell penetrating peptides��CPPs) ��ϸ��������(cell targeting peptides��CTPs)��ǰ���ܿ�Խϸ��Ĥת��ҩ������������Ե����ϸ���ϵ������ϡ�PDC ���Ķ��ױ�������ø���⣬�ʳ��÷�θ����;����ҩ ��PDC ��ѭ��ϵͳת�˲���ëϸѪ�ܱڵ����ϸ�������� CPP- ҩ��ż������ԣ���Ĥת����һ���������صĹ��̣�����ֱ�Ӵ���֬��˫���Ӳ� �����������ױ�����CPP- ҩ��ż�����ͨ��ת�����û�����鵼���������صķ�����ת��;������ϸ�� [32]������ CPPs �� [33] ��ת¼��ʽ�������� (trans-activator of transcription��TAT)��transportan��penetratin ������������������д�Ĥ�������ġ�CTP- ҩ��ż����Ŀ�Ĥת�������������������Ͻ鵼���������ã��ڴ˹�����ż����ͨ�����ں����ں��������ս�����ø�壬��������ͨ������ѭ���ص�ϸ��Ĥ���档���� CTPs ���������� - �ʰ��� - �Ŷ����� (RGD) �㶨ϵ���ġ������������ͷż��� (LHRH) �����ġ�ͨ���ɾ���չʾ����ɸѡ�������������������ĵ� [34]��
2.1.2 ϸ����ҩ��
���� PDC ż����ϸ����ҩ��ͨ���Ǿ���Ļ���ҩ�����ɼ�� (paclitaxel��PTX)��DOX��CTP �ȣ�����ͨ�����Ż����ϸ����ֳ���̶����ӿ��������ã�����ѡ���Բ��ߡ������������������������ϸ������֯�����ˡ��γ� PDC ��������Щҩ���������֯�İ����ԡ�������������֯�еķֲ����Ӷ��������Ӧ�����ƶ�����ҩ [35]��
2.1.3 ������
�����������Ӷ��ĺ�ҩ�����Ч������һ���õ������Ӳ���Ӱ����Ļ�ҩ��Ĺ��ܣ��ͷ��ӡ��ʵ����ȡ����ʵ��ȶ��Ժͼ��������������ӵĹؼ����ء��� ADC ���ƣ�PDC �����ӷ�Ϊ���ɶ����ͺͿɶ����� ( ���� 1)��ͨ�����ò�ͬ�������ӣ��ɵ���ҩ����ͷţ�����DZ�ڲ�����Ӧ�ķ��ա�
2.2 �ٴ�Ӧ�ý�չ
2018 �� 1 ������ FDA ���� lutetium Lu-177 dotatate( ��Ʒ�� Lutathera) ע��Һ���У��� PDC �ɷ���ҩ�� 177Lu ͨ����������ϼ� 1,4,7,10- �ĵ��ӻ�ʮ���� -1,4,7,10- ������ (DOTA) ����������ϣ���������������������ϸ�� ( ����������������������ԵĶ������� ) ��Ϻ��ڻ������� 177Lu �Ħ� ����ͨ����ϸ�����ڽ�ϸ�����γ����ɻ����յ�ϸ�����ˣ������������������������Ե�θ���������ڷ������� [36]�����Ƿ���ҩ���״α��������������ڷ���������Ҳ������ΪֹΨһ�������е� PDC��Ϊ���� PDC �ķ�չ�춨�˻��������ִ����ٴ��о��ε� PDC ҩ����� 3 ��ʾ [37��46]��

PTX ��һ�ֹ㷺�������Ƹ���������ϸ����ҩ������ٴ�Ӧ�ô���������ѡ������������ж����� -PTX ż���ﱻ�������ڸ��� PTX ���ܽ�ȺͿ˷� PTX �Ķ�ҩ��ҩ��ANG1005(Angiochem ��˾ ) ��һ���µ���ɼ�������ϵ�� 3 �� PTX ���ӹ������ӵ� angiopep-2 ���ϣ�ͨ����ϸ���������� LRP1 ��ϣ���ԽѪ - �����Ϻ�����ϸ�� [47]�������ٴ�������� ��ANG1005 ������ȷ�����ٰ���ת�����������������ٴ�����Ч������ANG1005 ���Ƶ����ٰ���ת�ƻ����ں�����ٴ������ʷֱ�Ϊ 77���� 86�������ٰ���ת��ͬʱ��������Ĥ�������� ANG1005 ���ƺ��������ӳ�����λ��������Ϊ 8 ���£����������κ����Ƶ����ٰ���ת�ƻ��ߵĹ���������Լ 2 ���£����û������Ʒ����Ļ��߹���������Ϊ 3 �� 4 ���¡�ANG1005 ���� HER2 �������ٰ����ߵĽ���������� HER2 �������ٰ����ߣ��� 2 ������Ⱥ�徭ANG1005 ���ƺ����λ�������ھ�����ʷ���ݸ�1 �����ϣ����� ANG1005 �İ�ȫ�����Ը��ƣ�������ȫ��������Ӧ [48]�������CHEN ��ɸѡ����ͬʱ��ԽѪ - �����Ϻ�Ѫ - ���������ϵĶ��� M1-RGD������ͨ���� ANG1005 ���Ƶķ����� PTX ���Ӻ�����Ч�ӳ���ԭλ U87 �Խ�����ģ��С��������ڣ��뽺�����ٴ�һ����ҩ��Ī������ʹ�ú�ɴ���ӳ�����С�������ڣ�Ϊ PTX �����Բ������������ṩ���о����� [49]��
���� PDC �������Ե��ٴ�Ӧ�����ƣ���Ŀǰ�������ڻ��Ƶ� PDC �������С����������ݶ��ԣ�PDC Ӧ�����ٴ����ܴ��ڵ��ϰ����� ���� PDC ����Է���������С���������ױ����������ȫ����ҩʱ��Ч������ ���� PDC �����е��������� L �Ͱ����ṹ�����ױ�Ѫ�е�ø��������� PDC �İ������ܱ����� ���� PDC Я��ҩ��������Ϳ�Խ����������ϵ���Ч���д���ߣ�����ϸ���� PDC��Ӧ����Ҳ�д������о���
3 �������� - ҩ��ż���� (ApDC)
����������ͨ��ָ������������ϵͳ����( systematic evolution of ligands by exponential enrichment��SELEX) �����ӵ����Ѻ������Ŀ���ɸѡ��õģ�����б�������ԡ���������ϵĺ��������� [50]���ڿ������ڰ������Ƶ� ApDC ����Ҳ�о�Ӧ��DZ�������������ͨ���������۵��γ��ض�����ά���� ( �緢�С������Ľǻ��� )����ͨ�����»�����������������á�����ѻ�������б��ϣ���������ƿ��� - ��ԭ�Ľ�ϣ�������������ֱ���Ϊ����ѧ���塱[51]�������뿹�幦�����ƣ������������Ծ����������ص����ƣ����Ľ�����������������൱����ߡ����⣬�뿹����ȣ���������������С�������ɱ��͡���ѧ���Ρ�����ԭ�Ե͡����μ����С����ѧ�ȶ��Ըߡ���֯���ٶȿ졢���Ե��ŵ� [52]������������ĺ���������������ڶ�б꣬��С���ӡ����ġ������ʣ���������ϸ���� [53]�������SEFAH��������ϸ��Ϊ�е㣬������һ�ּ�Ч�Ļ���ϸ���ĺ�������ѡ����� cell-SELEX��ʹ�����ϸ��Ĥ�������Ȼ����ѡ����������Ϊ���ܣ�ͨ�� cell-SELEX ѡ��ĺ���������Ϊ����ҩ����͵ĸ�����������������õ�Ӧ��ǰ�� [54]���� ADC ���ƣ�ApDC ͨ������������� ���������塢�����Ӻ�ҩ�� [55]�����к���������Ϊʶ�����壬���н�����ҩ����͵�Ŀ�겿λ�����Ŀ��������������ѧ���ܵ����á�Ŀǰ�����������������Ƶĺ���������� 4 ��ʾ [56��67]�����ں�������Ļ�ѧ�ȶ��Ըߡ���ѧ���μ����еķ��ӹ��̼���ʹ���������Ʒ� (���ơ�����Ʒ������ء������Ʒ�������� ) �������ϺͿɱ�� [68]��ApDC����ϸ��Ϊ���Ӽ� ApDC ������ ApDC��ǰ������������ϸ����ҩ��ͨ�����ۻ�ǹ�����ʽ������һ���Ե���������ʽ���� ��������ָ����������ϸ����ҩ�����Ӻ��ͨ��ijЩ���������������װ���Ӷ��γɾ���һ�������������ṹ��������Ҫ���۷��Ӽ� ApDC��

3.1 �ǹ������ӵ� ApDC
DNA ��һ���������ӣ���ͨ�����������װ��˫�����ṹ��ͨ���������-�� �ѻ�����ˮ����ñ����ȶ� [69]���ʿ�ͨ�����ӽ��Ͳ����ʵ��ҩ������롣cell-SELEX �ĺ���ԭ�����ڶз�������δ֪������£�ɸѡ�õ���ʶ������ϸ���ĺ������壬ֱ��������������������ơ�YOON��������һ���ԣ�ɸѡ�õ�һ�ֿɰ������ٵ����ٰ��ĺ������� P19����֤�������͵��ͼ���������������� (FU) �������������� DM1 ����������Щ����ҩ����̹Ѻ������ϣ�Ȼ��ͨ���ӽ�����������ϣ����ɵ� ApDC �ɽ�ϸ����ҩ�������Եص���������ϸ�������ٷ������Բ�����Ӧ [70]��DOX ��һ�ֳ��õĹ�������ҩ������һ��ƽ���Ļ��ṹ����Ƕ�� DNA �еļ��Ƭ�� ( �ر��� CG�� GC ����� ) �У����� DNA �ĸ��ƹ��� [71]������ DOX ��Ƕ�����ṹ��һ���ԣ��ɽ� DOX Ƕ�븻�� CG��GC ���еĺ��������У����� DOX ������͡��������� A10 ���� 71 ����������ɵĵ��� RNA��������ǰ���ٰ�ϸ������������ PSMA���������Խ�ϡ�BAGALKOT ��ͨ���ǹ�������ý� DOX Ƕ�� A10 ����ά�ṹ�еõ��� A10-DOX ż����ɽ� DOX �������͵� PSMA ���������ǰ���ٰ�ϸ�������� A10 �� DOX ͨ���ǹ��۷�ʽ��ϣ�A10 �� DOX ���ɱ��ָ�������ԣ���Ӱ����Թ�Ч�ķ��� [72]��Ϊ�����ҩ�����Ч�ʣ�ZHU ��ͨ���ӽ���ʽ��Ӧ���ں���������ӵ� 5'�������� 1 ���ɼ��� 100��ҩ�����λ����ɵij�˫�� DNA���乤��ԭ�������ڽ�����������Ϊ���������г�ʻ��Ŀ������ϸ���Ļ�ͷ��ͬʱ���������䡱��Ϊҩ��ĸ�Ч���壬��ҩ��ԴԴ���ϵ����͵���ϸ��������ѡ����ϸ������Ч�� [73]����ʾ������Ŀ�������Ч���������˲�����Ӧ���������õ�Ӧ��ǰ����
3.2 �������ӵ� ApDC
��Ȼͨ���ǹ��۽���γ� ApDC ���Ʊ������Ƚϼ�������ҩ�ﲢ������ЧǶ����������У�����ҩ���Ƕ����ܻ�ı��������Ľṹ���Ӷ�Ӱ�����������б�������Խ�ϡ���ˣ����˷ǹ��۽���⣬���۽��Ҳ���㷺���ڿ������ȶ�������λ������������DZ���� ApDC���ں���������ҩ���ϵĹ����У�ͨ����Ʋ�ͬ�������ӿ�ʹҩ�����ض�����֯����ϸ�������ͷš����磬������ҩ DOX �� DNA �������� sgc8( ���뼱���ܰ�ϸ����Ѫ�� T �ܰ�ϸ������������ PTK7 �����Խ�� ) �����γ� ApDC��������ȶ��������Ϊ�����ӣ����������������������Ժ��������ø�����ͷ� DOX�������������� [74]��LI �ȱ���һ����֯����ø B ���еĺ˵������� (NucA) -PTX ż�����ѡ���Եؽ� PTX ���͵�������λ�����������PTX �Ŀ��������Բ�������������Զ��� [75]�����ö���֯����ø B ���е� VC ���ļ��� NucA ���ӵ� PTX �� 2' λ�����ǻ��ϣ��õ������� NucAPTX ż��������ѭ�����ܱ����ȶ�����ż�����ϵ�NucA �ɴٽ�����������֯�еĻ��� ����������ϸ����NucA-PTX ż����Ķ��������ӱ�ø�⣬�ͷų� PTX ���������á������HE �Ƚ������������Ժ������� AS1411 �����ټ��ؽ���ż��������ż������������ʶ������ϸ������������ϸ����GSH �ļ����´���һϵ������������Ӧ��ԭλ����ѭ������̼�������ɻ� [76]��ͬʱ����ż����ļ�����������������ϸ���� GSH �ĺ�����ͬʱ��������������ĺ���������Эͬ�Ļ�ѧ����ѧ�Ʒ�ЧӦ����ż����������ٰ�ϸ�� (MDA-MB-231) ����ǿ�����Ժ�ϸ�����ԣ����ڿ����������ٰ���Ч���á��Խ�����֯�IJ�����ӦС��Ϊ���������ҩϵͳ����ƺ����ɻ���ط��ӻ��Ƶ��о��ṩ���¼��⡣
���ܺ�������������������ԣ��о�Ҳ֤���� ApDC ���������������Ӧ��DZ����Ȼ����������Ƶĺ�������� ApDC �Ŀ����Խ�Ϊ�ͺ�����Ϊֹ��ֻ��һ���Ժ�������Ϊ������ҩ��pegaptanib[ ��Ʒ�� Macugen��һ�־��Ҷ��� (PEG)���� VEGF �������壬�������������Իư߱��� ]������� FDA ������ [ 77] �������������Ƶĺ������� AS1411 Ŀǰ���ڢ����ٴ��о��� [78]��ApDC��ʵ���������ٴ�����˷�һϵ�����⣬���������ɸѡ���̽ϳ����ɹ��ʻ��ϵ� �����������ApDC ���ȶ��Խϲ�������ױ��ձ���ڵĺ���ø���⣬���ڰ�˥�ڽ϶̣����뾭һ���Ļ�ѧ���β��������ٴ� ������ϵͳ�Ժ����ʶ������������߷�Ӧ�ȡ�
4 �ۺ��� - ҩ��ż����
�ۺ��� - ҩ��ż��������һ�ֻ����ҩ��ͨ�����۽�����ӵ��ۺ����϶����ɵľ���ҩ�����ԵĴ���ӽṹ�����е�ҩ�������С����ҩ�Ҳ�����Ƕ��ġ������ʻ�������� [79]��ҩ����ۺ���Ľ���ж����洦����������ҩ����ܽ�ȡ�������ҩ�ٶȡ����ҩЧ����ҩ������ѧ��Ϊ�� [80]�������������žۺ���ż���������ս����죬Խ��Խ��ľۺ��� - ҩ��ż����������ٴ��о�����ʾ�����õķ�չǰ����
4.1 �������
�ۺ��� - ҩ�ﹲ��ż����ĸ���������RINGSDORF ������ 1975 ��������������Ϊ�ϳɾۺ���ҩ���ҩ�����Ծۺ��� [81]������ҩ����ۺ�������ͨ�����ȶ��������γɵģ��� 1 ���ۺ������ɺ� 3 ����ͬ�ĵ�Ԫ��ɣ���һ������ˮ����ʹ��������ӿ��ܺ��� ���ڶ�����ҩ����ۺ��������ӵ�����ҩ��ͨ��ͨ�������� ( ��������ż�����ġ�������� ) ��ϵ��ۺ��������ϣ������������ض������»���ѣ�ʹҩ��Ӿۺ����������ͷţ���ʹ�õ����������ͻ�Ӱ��ҩ��ż���������ϵı�����ҩ���ȶ��Ժ�ҩ���ͷŻ��� ����������ʵ�ְ�������������书���ǽ������ۺ�����ϵ���͵�Ŀ��ϸ����ҩ�����ã���Ŀǰ�о��й㷺Ӧ�õİ�������������ơ�ͨ���Դ���Ӿۺ������IJ�ͬ������в�ͬ����ƿ�ʵ�ֲ�ͬ�Ĺ��ܡ�
Ŀǰ������ҩ����͵ľۺ�����Ҫ���� [82] ���پ۰������������� L- ������ (PLL)���� L-�Ȱ��� (PGA)���������� [N-(2- ���һ� -L- �Ȱ����� )](PHEG) �;��Ŷ����� (PASP) �� ���ڶ�Ԫ�ᣬ��� �� ƻ���� (poly-��-malic acid��PAMA)���� ��ƻ���� (poly-��-malic acid��PBMA) ���۶��ǣ����Ͼ��ǡ���³�����ǡ������ᡢ�Ǿ��� ������������ N-(2- �DZ��� ) ����ϩ���� (HPMA) �������PEG �ȡ�һЩ���������ڽ��컷�ࡢ�������ɼ�����С���ӻ���ҩż�����ۺ����ϡ��㷺����ҩ��ֱ��ż����ͨ����������ۺ�����ż���Ĺ������а������Ȼ����ǻ����ϻ��� [83]���ۺ��� - ҩ��ż����ɷ�Ϊ�ۺ��� - �����ż����ۺ��� - С����ż�����֦״����Ӻ;ۺ��������� [84]��������Ҫ��ע�������������Ƶľۺ��� - ҩ��ż����ǰҩ��
4.2 �ٴ�����
�����ڿ������ľۺ��� - ҩ��ż���� [ ��PGA���Ա��������� (PDM) �������������Ӷ��� ] ������ 1975�꣬�ýṹ�оͰ��������� 3����Ԫ [85]��MATSUMURA �ȱ���һ�ֿ������������εľۺ���ż���ᄇ��ע��������ȸ�����������֯�У����������ǿ���������� (EPR)ЧӦ[86]��������ҩ��ľۺ���ż�������ٴ�ǰ����ģ���б��ֳ����ߵİ�ȫ�Ժ���Ч�ԣ�����ҩ�ﶼ�������ٴ�����Σ������Ѿ��������� ( ���� 5)[87��96]��

PK1 �ǵ�һ�������ٴ��о���ˮ���Ծۺ��� -С����ҩ��ż���ͨ����ø����ѽ������(GPLG) �� DOX �� HPMA �������� [ 97]��PK2�� PK1 ���ƣ������п���ۺ�������ϵİ����ǰ�����˿�����ΰ�ϸ���ϵ�����Һ���ǵ���(ASGP) ���� [98]�������ٴ�����������������Ի���ҩ���������У�PK1 ��������˥��Ϊ 93 h�������� DOX ��ѭ��ʱ�������ӳ�������Ը�С [99]��PK1 �Ģ����ٴ�������ʾ�����ٰ� (7/62)����Сϸ���ΰ� (16/62) �ͽ�ֱ���� (29/62) ����ÿ 3 ��ʹ�� 280 mg/m2 �ļ������� 6 �����߳��ֲ��ַ�Ӧ��Ȼ��������һС���ֻ����й۲쵽 PK1 �������и������������ӳ��� DOX ��ѭ��ʱ�䡢���������ڰ�ȫ�ԣ�����Ч������ [100]���ź����ǣ��ڢ����ٴ������У�2 �����ֳ� PK1 ��������ЧӦ�Ļ��߶�����û�з�Ӧ�����������з�Ӧ�Ļ���ȴδ�۲쵽����������������������ʾ��PK1 �����������Dz����ȵģ���������ʵ������Ч�������ǵ������������һ����������Ʒ�Ӧ����ʹ�����Ժõ�������Ҳ������Ч�������� PK1 �İ�˥��û���������ӣ��������Ҳ�����룬�����俪��ʧ�� [101]��
PGA-PTX(Opaxio��ԭ�� Xyotax) ���������Ŀ��������ö����㷺�о������ξ���ע���ҩ�����ȫ����С�����ٰ�����δż���� PTX ��ȣ���ż�������������������� 2 ������������������� 12 �����뵥�ξ���ע����ȣ����ע���ż����������Ƶ���Ч��������ѭ��ҩ��ż���� ( ��PGA-PTX) ����������θ�ҩ�ȶ�θ�ҩЧ������ [102]�����ǣ����ѳ������ߵĢ����ٴ������У���ż�����Ӧ���ʽ�Ϊ 10�� (10/99)����λ������Ϊ 2 ���� [103] ���ڷ�Сϸ���ΰ����ߵĢ����ٴ��о��У���ż�����黼�ߵ���������������൱�������ڷ�Сϸ���ΰ�һ�����Ʒ��� PTX/ ������ȣ�PGA-PTX/ �������ṩ���ѵ������� [104]������PGA-PTX ������ PTX ���ܽ�ȼ���ȫ�ԣ������ٴ���������Ч����ĸĽ���Ȼ���ޡ�
ϸ����ҩ�������濵���ı� PEG ż����(Onzeald) �ѽ�������ٴ����顣Onzeald ���ÿ��ѽ��������ÿ�� PEG ���Ͻ�� 1 �������濵���ӣ���Է�������Ϊ 20 000 �������ڣ���������ˮ���ͷų������濵������л�ɻ��Կ������ɷ�SN-38[105]���ڶ���ģ���У��봫ͳ�������濵��ȣ�Onzeald ���ֳ��ӳ���ѭ����˥�ڡ���Ѫ�����������ȶ���Ũ�Ⱥߴ� 400 ����Ѫ����¶ (AUC)���������濵��ȣ�ʹ�� Onzeald �������е� cmax ������ 10 ������Ѫ���е� cmax ȴ�����ˣ���ʾ������ָ���������ƣ��ʸ��� Onzeald �ɵ��³������ܵ��������ƺ��������ˡ��ڢ����ٴ������У�Onzeald֤ʵ�����Ƶ�ҩ������ѧ������SN38 ���ֳ� 50 d��������˥�ڣ��������濵��ҩ���˥��ֻ�� 12 ��47 h[106]��Onzeald ������ٴ��������鲢�������У��������ư�����ת�Ƶ����ٰ� [107]��
�������еľۺ��� - ҩ��ż�����Ʒ�����PEG- ������ż����������Ʊ����ס������ܰ�ϸ����Ѫ�������ʪ�ؽ��� [80]�����֮�£�С����ҩ��ľۺ���ż������ٴ�Ӧ��һֱ�����ޣ�Ŀǰֻ�� PEG- ����ͪż���� ( ��Ʒ��Movantik) �ɹ������г�����������������ʹ���߰�Ƭ��ҩ������ı��� [ 108]������һЩ�ۺ���ż������֤�����ӳ���˥�ںͽ��Ͷ��ԣ����ڿ�������Ч����ĸĽ���Ȼ���ޡ��������ۺ����Ʒ����ٴ�ǰ��Ч�ںܴ�̶��Ϲ����� EPR ЧӦ�鵼���������ۣ������������еı�������Ŀǰ��һ����������Ļ��⡣���磬37 ������ PK1 ���Ʋ����Т��ں͢����ٴ����������Ļ����У�ֻ�� 8 ��ͨ�������Ժ��س�����ʾ��������ȡ [ 109]���ٴ�ǰ�о��ͻ�������֮����ֲ���ԭ�������С��ģ�Ͳ���ȷ�ط�ӳ�����������ص㣬С�������Ŀ����������²������Ѫ���γɺ�Ѫ����©����������������������Ѫ�ܶ�����©������һ���о����������ͨ�� EPR ЧӦ�鵼������ҩ��������������У�ֻ�� 0.7������ע�������ҩ�ﵽ������ [110]������ʾ ����ʹ���ٴ�ǰģ���б��ֳ���������Ч����ҩ�����壬����������λ���͵�ҩ��Ҳ�Dz���ġ�Ȼ���о��߲�û�ж�����С����ҩ���������λ���з������Ӷ�������ҩ�������С����ҩ����������λ�����Ч������ˣ�δ����Ҫ��ϸѡȡ���ߣ��Ա�ȷ����Щ���������������趨���Ʒ� [111]�����߿������Ӿۺ���������������ķ��� ( �������������� )[112]��ͬʱ�����ҩ������ѧ������ֲ������������ٴ�ǰ��������ȷ����ѡ��������������ھ����㹻���ȶ��ԣ���֤ҩ�����Ч���͡�
5 �ܽ���չ��
������Ҫ�����˿��塢���ġ���������;ۺ�����ҩ���ż�����ڽ鵼ҩ���������е�Ӧ�ã���Ŀǰ�о����ٴ����������ԣ����Ǿ������õ�Ӧ��ǰ�������ֲ�Ʒ�����ٴ�Ӧ����ȡ�������õ�����Ч����Ϊ���ߴ����˽ϴ��洦����ͬʱҲ������һЩ���ֵ����⣬���粻����Ӧ����ҩ�ԡ����ð���ȱ�ٽϺõ����������ȡ����ȣ�����������Ȼ�����ҩ����������λ�����������������֯�еķֲ��Բ��ɱ��⣬���²���������Ӧ�����磬Kadcyla �IJ�����Ӧ��Ȼ�Ȼ����������ᣬ���Դ��ڶ��ġ�Ż�¡�ѪС����١����������ʹ�Լ��ζ��ԵȲ�����Ӧ�����»�����ҩ˳Ӧ�Խϲ��Σ��봫ͳ����ҩ��һ������������Ҳ������ҩ���⣬����ϸ������߱����ҩ�����ŵ��ף��ɴٽ�ҩ�����ţ���������������չ�����п��ܳ��ָ��ӵ�ͻ�����ٽ�������������ʹ����ҩ��ʧȥ���á������������������ƵĻ��ѽϰ��������ǿ�����ҩ��������ƹ����л�����һЩ�������ã��ںܴ�̶������������������ߵľ��ø�����������������ĸ����Լ������ԣ����²�ͬ�������ߵ���Ч���ڲ��죬��Ҫ��һ��Ѱ�ҿ���Ԥ�����Ч��������������ɸѡ�����ʺϰ������ƵĻ��ߣ�ʵ�־����ơ�������������ҽҩ�Ƽ��Ľ����Ͱ����ҩ���۵����ƣ�����ٽ���Ϊ������Ч�İ���ҩ����ٴ�ת����Ϊ����������ߵ����ƴ���������
��������������Ϊ��ҵ����ѧϰ����Ȩ��ԭ����ԭ��־���У�������Ȩ������ϵɾ�������±�ע���������³����������Ķ�ԭ�ļ��ο����ף����Ķ�ԭ��־��
