
№эИҐµД¶юК®ДкЈ¬ЦЧБцЦОБЖТСѕґУ·ЗМШТмРФµД»ЇБЖПтѕЯУРСЎФсРФµД»щУЪ·ўІЎ»ъЦЖµДБЖ·ЁЧЄ±д[1]ЎЈ·ЦЧУ°РПтЦОБЖЧчОЄЦЧБцЦОБЖµДРВКЦ¶ОЈ¬ХэТФЖдБЖР§ёЯЎўІ»Бј·ґУ¦ЙЩЗТЗбµИМШµг¶ш±ёКЬЦхДїЈ¬ПЦТСіЙОЄЦЧБцЦОБЖБмУтµДСРѕїИИµгЎЈЛьТФЦЧБцОў»·ѕіЎўЦЧБцПё°ыПё°ыД¤»тПё°ыДЪМШТмРФ±нґп»тёЯ±нґпµД·ЦЧУОЄЧчУðе㣬ДЬ№»ёьјУМШТмµШЧчУГУЪЦЧБцПё°ыЈ¬Чи¶ПЖд¶сРФФцЦіЎўЧЄТЖ»тУХµјЖдµтНцЈ¬Н¬К±ЅµµНБЛ¶ФХэіЈПё°ыµДЙ±ЙЛЧчУГЈ¬ТтґЛКЗК®·ЦУРЗ°НѕµДЦЧБцЦОБЖ·Ѕ·ЁЦ®Т»[2]ЎЈ
ЦОБЖЦЧБцµДЙъОпЦЖјБЦчТЄ°ьАЁµҐїЛВЎї№МеЎўµ°°ЧЦКєНлДЎЈ¶ФУЪЎ°РЎРН»ЇЎ±µДлДАа¶шСФЈ¬ЖдєПіЙ№¤ТХіЙКмОИ¶ЁЎўІ»ТЧФЪМеДЪУХ·ўГвТЯУ¦ґрЎўѕЯУРБјєГµДЦЧБцґ©НёДЬБ¦ТФј°ЙъОпјжИЭРФЈ¬ПФКѕБЛБјєГµДСРѕїјЫЦµєНУ¦УГЗ°ѕ°[3-5]ЎЈДїЗ°ТСЙПКРµДлДАаТ©ОпУР60ёцЧуУТЈ¬ЖдЦРПъКЫ¶оі¬№э10ТЪГАФЄµД4ёцлДАаТ©ОпЦРУР3ёцЈЁББ±ыИрБЦЎўёкЙбИрБЦєН°ВЗълДЈ©УГУЪЦЧБцµДЦОБЖ»тЦЧБцІў·ўЦўµДёЁЦъЦОБЖЎЈ2000ЁC2010ДкЈ¬ЅшИлБЩґІКФСйµДТФ¶аЦЦРОКЅУГУЪЦЧБцХп¶ПєНЦОБЖµДлДАаТ©ОпХјХыёцлДАаТ©ОпµД18%Ј¬ЖдФЪї№ЦЧБцТ©ОпЦР·ў»УЧЕФЅАґФЅЦШТЄµДЧчУГ[3]ЎЈ°РПтлДІ»µ«їЙТФЧчОЄј¤¶ЇјБ»тЮЧї№јБУГУЪЦЧБцµДЦОБЖЈ¬»№їЙТФЧчОЄПё°ы¶ѕТ©ОпЎў·ЕЙдРФєЛЛШЎўПФПсКФјБТФј°РВРНЦЖјБЈЁДЙГЧЎўЦ¬ЦКМеЎўЅєКшµИЦЖјБЈ©µИµД°РПтґ«КдФШМеЈ¬ОЄЦЧБцµДХп¶ПєНЦОБЖїЄ±ЩБЛРВµДНѕѕ¶ЈЁНј1Ј©ЎЈ±ѕОДѕН»щУЪї№Меј°ЕдМеµДЦЧБц°РПтлДТ©ОпСРѕїЅшХ№ЧчјтТЄЧЫКцЎЈ
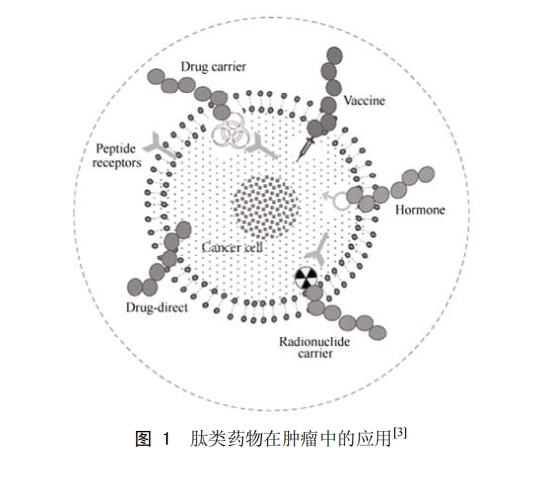
1 »щУЪї№МеµДЦЧБц°РПтлДТ©Оп
ї№Ме·ЦЧУЦШБґєНЗбБґµДїЙ±дЗшЦРЈ¬ёчУР3ёцЗшУтµД°±»щЛбРтБРёЯ¶И±д»ЇЈ¬іЖОЄёЯ±дЗшЈЁhypervariableregionЈ¬HVRЈ©»т»ҐІ№ѕц¶ЁЗшЈЁcomplementaritydeterminingregionЈ¬CDRЈ©Ј¬·Ц±рОЄCDR-H1ЎўCDR-H2ЎўCDR-H3єНCDR-L1ЎўCDR-L2ЎўCDR-L3Ј¬ЛьГЗ№ІН¬РОіЙТ»ёцДЬ№»К¶±рј°ЅбєПї№ФµДИэО¬±нГ滥І№ЗшЈЁНј2Ј©ЎЈCDRsѕц¶ЁЧЕї№МеµДМШТмРФУлЗЧєНБ¦Ј¬ФЪї№ФЅбєПЦРЖрЧЕ№ШјьЧчУГЎЈСРѕї±нГчЈ¬ї№МеµДМШТмРФУЙОЄКэІ»¶аµД°±»щЛбІР»щѕц¶ЁЈ¬ХвОЄ»щУЪї№МеCDRsµДлД¶О±ЈБфНкХыї№МеµДЅбєП»оРФєНЙъОпС§№¦ДЬМṩБЛїЙДЬЈ¬ОЄї№МеµДРЎРН»Їµм¶ЁБЛБјєГµДАнВЫ»щґЎ[6-7]ЎЈ
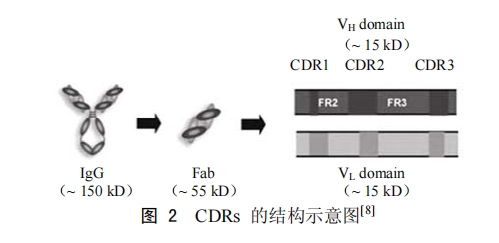
1.1 АґФґУЪї№МеCDRsµДЦЧБц°РПтлДТ©Оп
АґФґУЪї№МеCDRsµДCDR-H3РтБРТІіЖЎ°ОўЈЁРЎЈ©ї№МеЈЁmicro/miniantibodyЈ©Ў±Ј¬ЛьєННкХыї№МеѕЯУРАаЛЖµДМШТмРФЎЈCDR-H3О»УЪї№ФК¶±рО»µгµДЦРРДО»ЦГЈ¬ФЪГвТЯ·ґУ¦ЦР·ў»УЧЕЦШТЄЧчУГЈ¬КЈПВµДCDRsФтО§ИЖЖдЦЬО§ТФЅшТ»ІЅФцјУЖдЗЧєНБ¦ЎЈЧоЅьСРѕї±нГчЈ¬іэБЛCDR-H3ТФНвЈ¬ЖдЛыµҐ¶АµДCDRsТІїЙДЬНЁ№эПИМмГвТЯ·ґУ¦·ЦЧУАґ·ў»УЙъОп»оРФ[7]ЎЈC7L1ЎўC7L2ЎўC7L3ЎўC7H1ЎўC7H2єНC7H3КЗСЬЙъУЪµҐїЛВЎї№МеC7ЈЁї№°ЧЙ«ДоЦйѕъПё°ы±ЪёКВ¶МЗµ°°Чї№МеЈ©CDRs6ёцЗшУтµД6ЦЦлД¶ОЈ¬Blast±И¶ФЅб№ы±нГчЛьГЗєНЙъОпМе·ЦГЪµДМмИ»лДАаµД°±»щЛбРтБРґжФЪєЬґуІоТмЎЈХвР©лД¶Оѕ№эТ»ПµБРМеНвМеДЪІвКФєуЈ¬·ўПЦ»щУЪCDR-H2µДC7H2лДЈЁYISCYNGATSYNQKFKЈ©ѕЯУРЧојСµДї№ЦЧБцР§№ыЎЈЛьДЬПФЦшТЦЦЖКуєЪЙ«ЛШБцПё°ыB16F10µДЙъі¤ЈЁIC50ЦµОЄ50¦Мmol/LЈ©ЎўТэЖрDNAµДЅµЅвЈ¬ґЛНв»№їЙµјЦВєЪЙ«ЛШБцПё°ыТФј°°ЧСЄІЎПё°ыµДµтНцЈЁcaspaseТААµРНЈ©Ј¬ЖдµтНц»оРФУллДµДКЬМеЅбєП»оРФУР№Ш[7]ЎЈєуРшСРѕї±нГчЈ¬C7H2µД·ЦЧУ°РµгОЄ¦В-јЎ¶Їµ°°ЧЈ¬ЛьїЙУХµј¦В-јЎ¶Їµ°°ЧµДѕЫєПЎўF-јЎ¶Їµ°°ЧµДОИ¶ЁТФј°ЦЧБцПё°ыµДЛАНцЈ¬¶шЗТїЙТФТЦЦЖЦЧБцµДЧЄТЖЈ¬µ«¶ФХэіЈПё°ыТФј°РЎКуОЮГчПФµД¶ѕРФ[9]ЎЈ
Н¬К±АґЧФУЪCDRsµДлД¶ОЈ¬ЖдМеДЪЙъОп»оРФµДУРОЮїЙДЬУлCDRµДСЎФсТФј°УРОЮјдёфЗшУР№ШЎЈОЄБЛИ·¶ЁЧојСµДЕд¶Ф·ЅКЅЈ¬УРИЛЅ«ї№EBІЎ¶ѕ°ьД¤ї№ФµДHB-168ї№МеCDR1»тCDR2УлCDR3НЁ№эCDRТФНвЗшУтµД№ЗјЬЗш2ЈЁframeworkregionЈ¬FR2Ј©Б¬ЅУЖрАґЈ¬РОіЙІ»Н¬ЧйєПµДДвлДЈ¬ЖдЦРТФCDRH1-FRH2-CDRL3ЈЁє¬28ёц°±»щЛбЈ©РОКЅЧйєПµДДвлДµДї№ФЅбєП»оРФЧоёЯЈ¬ЗТЖдґ©НёІўё»јЇУЪКµМеБцµДДЬБ¦±ИНкХыї№МеЗїЈ»¶шОЮјдёфЗшµДДвлДЈ¬ИзCDR H1-CDRL3Ўў6ёцCDRsРОіЙµД»·лДµИЈ¬УЙУЪИ±·¦Ў°ЧјЙъАнС§Ў±јдёфЗшЈ¬ЛьГЗУлї№Ф±нО»ЅзГжµДЅбєПУРЛщјхИхЎЈУЙґЛїЙјыНЁ№эјдёфЗшБ¬ЅУµДCDRsёьјУЅУЅьМмИ»ї№МеCDRµД№№РНЈ¬¶шЗТґ¦УЪ¶ФЅЗПЯО»ЦГµДCDR1єНCDR3¶Фї№ФµДК¶±рЖрЧЕЦШТЄµДЧчУГ[10-11]ЎЈ
1.2 ДЈДвї№МеєНКЬМеП໥ЧчУГµДЦЧБц°РПтлДТ©Оп
±нЖ¤Йъі¤ТтЧУКЬМе2ЈЁhumanepithelialgrowthfactorreceptor2Ј¬HER2/ErbB2Ј©КЗ±нЖ¤Йъі¤ТтЧУјТЧеіЙФ±Ц®Т»Ј¬УлЖдЛы3ёцјТЧеіЙФ±І»Н¬Ј¬HER2Г»УРУлЦ®¶ФУ¦µДЕдМеЈ¬ЗТТФН¬Фґ»тТмФґ¶юѕЫМеРОКЅ·ў»УЧчУГЎЈHER2µД±нґпЛ®ЖЅУлИйПЩ°©µДЙъі¤ЎўЧЄТЖј°Ф¤єуіКПЦГчПФµДХэПа№ШРФЎЈЕБНЧЦ鵥ї№ЈЁУГУЪЦОБЖHER2СфРФЧЄТЖРФИйПЩ°©µДИЛФґ»ЇµҐїЛВЎї№МеЈ©ДвлДHRAPЈЁAc-PHAHF-NH2Ј©КЗКЧґОАыУГ»щУЪЅб№№µДТ©ОпЙијЖЈЁstructure-baseddrugdesignЈ¬SBDDЈ©ИнјюДЈДвЕБНЧЦ鵥ї№єНHER2µДЅбєПО»µгЈ¬ІўНЁ№эУЕ»ЇµГµЅµДТ»МхїЙТФМШТмРФµШУлHER2¶юѕЫМ幦ДЬУтЅбєПµД¶МлДЎЈёГлДїЙНЁ№эТЦЦЖPTENєНAktµДБЧЛб»ЇАґТЦЦЖHER2№э¶И±нґпµДИйПЩ°©Пё°ыµДФцЦіЈ¬ґЛНвЛь»№їЙТФФцЗїЧПЙјґјµДПё°ыµтНцУХµј»оРФ[8,12]ЎЈ
лДТЯГзКЗлДАаТ©ОпСРѕїµДИИµгЦ®Т»Ј¬ЧоРВБЩґІКэѕЭ±нГчДїЗ°ЅшИлБЩґІКФСйµДлДАаТ©ОпґуІї·ЦОЄлДТЯГз[13]ЎЈMVF266 CycКЗ»щУЪЕБНЧЦ鵥ї№ЅбєПHER2°ыНвУтµДµҐѕ§Ѕб№№ДЈДвЙијЖµДТ»ЦЦЕБНЧЦ鵥ї№СщлДТЯГзЈ¬Жд¶ЇОпМеДЪБЖР§УлЕБНЧЦ鵥ї№ПаІоОЮјёЈ¬µ«ОЮї№МеіЈјыµДё±·ґУ¦ЎЈMVF597CycКЗАыУГН¬СщµД·Ѕ·ЁЙијЖµДлДТЯГзЈ¬ЖдЙъОп»оРФУлЗъНЧЦ鵥ї№АаЛЖЎЈБнНвТ»ЦЦТСЅшИлБЩґІIЖЪКФСйЈ¬УГУЪГвТЯБЖ·ЁµДлДТЯГз14633ФтКЗУЙЕБНЧЦ鵥ї№СщлД¶ОMVFHER-2[597-626] єНЕБНЧЦ鵥ї№СщлД¶ОMVFHER-2[266-296]БЅІї·ЦЧйіЙЈ¬їЙТФ°РПтHER-2°ыНвУтµДБЅёцІ»Н¬±нО»[14]ЎЈ
AHNPЈЁanti-HER2/neupeptidomimeticЈ©КЗТФї№HER2µҐїЛВЎї№Ме4D5ЈЁHerceptinЈ¬єХИыНЎЈ¬УГУЪЦОБЖHER2№э¶И±нґпµДЧЄТЖРФИйПЩ°©Ј©єНHER2ёґєПОпµДИэО¬Ѕб№№ОЄ»щґЎЙијЖіцµДТ»¶О»·НвРЎлДЈЁFCDGFYACYKDVЈ¬·ЦЧУБїОЄ1.5kDЈ©ЎЈAHNP¶юј¶Ѕб№№Улї№МеµДCDR3АаЛЖЈЁН¬ОЄ¦В-ЧЄЅЗЅб№№Ј©Ј¬ПтНвСУЙмµДКиЛ®РФОІ°НФтУлї№МеµДFRАаЛЖЈЁіКПЦ¦В-ХЫµюСщµДА©Х№Ѕб№№Ј©ЎЈМеНвЅб№ы±нГчAHNPєНHER2ѕЯУРєЬёЯµДЅбєПДЬБ¦Ј¬ЖдЗЧєНБ¦іЈКэKDЦµёЯґп300nmol/LЈ¬МеДЪЅб№ыФт±нГчAHNPїЙТФДЈДвї№Ме4D5 µД№¦ДЬЈ¬ґУ¶ш·ў»Уї№ЦЧБцБЖР§ЎЈґЛНвЈ¬AHNPУлЖдЛы»ЇБЖТ©ОпБЄУГїЙТФФцјУЛьГЗµДБЖР§ЎЈДїЗ°Ј¬AHNPЧчОЄТ»ЦЦРВУ±µДРЎ·ЦЧУМЅХлТС№г·єУГУЪБЩґІЗ°СРѕї[15-16]ЎЈ
AERPЈЁanti-EGFreceptorpeptidomimeticЈ©ФтКЗ»щУЪПаН¬ФАнЈ¬ТФї№МеC225µДCDR-H3ОЄДЈ°еЙијЖµДТ»¶Ої№EGFRК®БщлДЈЁYCASRDYDYDGRCYFDЈ©ЎЈAERPєНEGFRµДЅбєПіКПЦЕЁ¶ИТААµРФЈ¬ЖдKDОЄ400nmol/LЈ¬ѕ99mTc ±кјЗµДAERPїЙФЪЦЧБцІїО»МШТмРФё»јЇЈ¬ЖдЦЧБцУлСЄТєµДЕЁ¶И±ИКЗµҐБґї№МеЈЁsingle-chainantibodyfragmentЈ¬scFvЈ©µД3.2±¶Ј¬µ«І»ЧгµДКЗЛьФЪёОФаєНЙцФаТІУРґуБї·ЦІјЈ¬ЖдТ©ґъ¶ЇБ¦С§УРґэЅшТ»ІЅМбёЯ[16-17]ЎЈ
2 »щУЪЕдМеµДЦЧБц°РПтлДТ©Оп
ЧФґУ·ўПЦМШТмРФКЬМеТФАґЈ¬ЕдМеµДлДАаЛЖОпѕНТтОЄѕЯУРБјєГµДКЬМеЅбєП»оРФЎўµНГвТЯФРФТФј°¶АМШµДЙъОп¶ЇБ¦С§РРОЄ¶ш№г·єУГУЪ°©ЦўµДХп¶ПєНЦОБЖ[18-21]ЎЈ±ѕІї·ЦТФДїЗ°іЈјыµДКЬМеАґ¶Ф»щУЪЕдМеµДЦЧБц°РПтлДТ©ОпЅшРР·ЦАаЎЈ
2.1 ЧчУГУЪј¤ЛШКЬМе
ґЩ»ЖМеј¤ЛШКН·Еј¤ЛШЈЁluteinizinghormone-releasinghormoneЈ¬LHRHЈ©лДАај¤¶ЇјБКЗЦОБЖЗ°БРПЩ°©µДЧоѕµдµДАэЧУЎЈДїЗ°Ј¬ТСЙПКРµДј¤¶ЇјБУРЈєІјЙбИрБЦЎўёкЙбИрБЦЎўЧй°±ИрБЦЎўББ±ыИрБЦєНЗъЖХИрБЦЎЈХвР©ј¤¶ЇјБКЗLHRHµДАаЛЖОпЈ¬їЙТФПВµчґ№МеµДґЩ»ЖМеј¤ЛШКН·Еј¤ЛШКЬМеЈ¬ґУ¶шТЦЦЖґЩВСЕЭј¤ЛШЎўґЩ»ЖМеј¤ЛШТФј°ШєНиНЄµДКН·ЕЎЈЛжєу·ўПЦµДлДАаЮЧї№јБИз°ў°НИрїЛЎўОчЗъИрїЛЎўµШјУИрїЛµИЅшТ»ІЅМбёЯБЛј¤ЛШЧи¶ПБЖ·ЁµДБЖР§[3]ЎЈ
ЧФ20КАјН80ДкґъТФАґЈ¬ґуБїКэѕЭ±нГч¶аКэЦЧБцПё°ыТИµєЛШСщЙъі¤ТтЧУКЬМеЈЁinsulin-likegrowthfactor-1receptorsЈ¬IGF-1RЈ©іКПЦёЯ±нґпЎЈБчРРІЎС§СРѕїТІПФКѕСЄЗеIGF-1Л®ЖЅєН»јИйПЩ°©ј°З°БРПЩ°©µД·зПХПа№ШЎЈIGFРЕєЕПµНіФЪЦЧБц·ўЙъ·ўХ№ЦРЖрЧЕЦБ№ШЦШТЄµДЧчУГЈ¬IGF-1RЧчОЄ°РПтЦОБЖµД°Рµг±»№г·єСРѕї[22-23]ЎЈJB3ЈЁD-CSKAPKLPAAYCЈ©ЧчОЄIGF-1ЧоУРР§µДАаЛЖОпЈ¬їЙТФєНДЪФґРФЕдМеѕєХщРФЅбєПIGF-1RЈ¬ґУ¶шТЦЦЖДіР©ЦЧБцµДЙъі¤ЎЈJB3НЁ№э¶юБтјьРОіЙ12лДЈ¬ЖдЦР°ьє¬Т»ёцDРН°±»щЛбЈ¬їЙУРР§¶Фї№ГёµДЛ®ЅвЎЈјт»ЇJB3єуµГµЅµД»·4лДJB9ЈЁD-CSKCЈ©ТІїЙУРР§µШё»јЇУЪIGF-1RСфРФЦЧБцІїО»[24]ЎЈ
БнНвЈ¬УлЦЧБцУР№ШµДј¤ЛШКЬМе»№°ьАЁЙъі¤ТЦЛШКЬМеЈЁsomatostatinreceptorsЈ¬SSTRsЈ©ЎўСЄ№Ь»оРФі¦лДКЬМеЈЁvasoactiveintestinalpeptidereceptorsЈ¬VIPRsЈ©µИ[19]ЎЈ
2.2 ЧчУГУЪEGFR
±нЖ¤Йъі¤ТтЧУКЬМеЈЁepidermalgrowthfactorreceptorЈ¬EGFRЈ©ФЪРн¶аЦЧБцПё°ыЦР№эБї±нґпЈ¬УлЦЧБцПё°ыµДФцЦіЎў·Ц»ЇєНЧЄТЖµИ№ШПµГЬЗР[25]ЎЈ
EGFRµДМмИ»ЕдМеEGFѕЯУРєЬЗїµДЙъОп»оРФЈ¬їЙДЬТэЖрГвТЯФРФµИОКМвЈ¬ТтґЛ±ШРлС°ХТРВµД°РПт·ЦЧУЎЈEGFє¬3ёцУЙ¶юБтјьРОіЙµД»·ЧґЅб№№Ј¬ЖдЦРB»·УлEGFRЅбєПУР№ШЈ¬»оРФЗшРтБРОЄCMYIEALDKYACЎЈИЛ№¤єПіЙµД»·лДїЙТФ°РПтEGFR№э±нґпЦЧБцПё°ыЈ¬ІўїЙЧчОЄТ©ОпµДФШМеТФМбёЯ»ЇБЖТ©ОпµДї№ЦЧБцБЖР§ЎўЅµµНИ«ЙнРФ¶ѕРФ[26]ЎЈ
БнНвЈ¬ТІУРСРѕї±нГчEGFµДC-¶ЛФЪЕдМе-КЬМеЅбєП№эіМЦР·ў»УЧЕЦШТЄЧчУГ[27]ЎЈНЁ№э»щТт№¤іМјјКхЅ«EGFµДC»·22ёц°±»щЛбЈЁјтРґіЙEcЈ©УлБ¦ґпГ№ЛШµДёЁ»щµ°°ЧЈЁlidamycinapoproteinЈ¬LDPЈ©ИЪєПРОіЙµДИЪєПµ°°ЧEc-LDP ±гКЗТ»ёцєЬєГµДАэЦ¤ЎЈELISAєНПё°ыБчКЅјмІвЅб№ыѕщПФКѕEc-LDPµ°°Ч¶ФEGFRёЯ±нґпµДЦЧБцПё°ыПµA431єНMCF-7¶јУРєЬЗїµДГвТЯЅбєП»оРФЈ¬¶ш¶ФІ»±нґпEGFRµДNIH3T3Пё°ыФтОЮЅбєП»оРФЈ¬ГвТЯУ«№вКµСйТІЦ¤КµEc-LDP µ°°ЧїЙУлA431Пё°ыД¤КЬМеЅбєПЎЈФЪґЛ»щґЎЙП№№ЅЁµДЛ«°РПтИЪєПµ°°ЧEc-LDP-HrїЙН¬К±°РПтEGFRєНHER2Ј¬ЅшТ»ІЅЦ¤КµБЛEc№СлДµД°РПтРФ[28-29]ЎЈ
2.3 ЧчУГУЪХыєПЛШКЬМе
ХыєПЛШЈЁintegrinЈ©КЗУЙ¦БСЗ»щєН¦ВСЗ»щЧйіЙµДТмФґ¶юѕЫМеЈ¬ЛьКЗТ»АаПё°ы±нГжКЬМеЈ¬ЖдЕдМеКЗПё°ыНв»щЦКµ°°ЧЈ¬ИзЅєФµ°°ЧЎўПЛр¤Б¬µ°°ЧЎўІгр¤Б¬µ°°ЧЎўПё°ыјдр¤ёЅ·ЦЧУЎўСЄ№ЬПё°ыр¤ёЅ·ЦЧУµИ[30]ЎЈУ°ПмЦЧБцСЄ№ЬЙъіЙµДХыєПЛШЦчТЄ°ьАЁ4ЦЦЈє¦Бv¦В3Ўў¦Бv¦В5Ўў¦Б5¦В1єН¦Б2¦В1ЎЈЖдЦР¦Бv¦В3КЗЧоТэИЛ№ШЧўµДЦЧБцСЄ№ЬР°бк[31]ЎЈ
ХыєПЛШєНЕдМеµДК¶±р№эіМУлЕдМеµДМШ¶Ё°±»щЛбРтБРУР№ШЎЈ1997ДкЈ¬PasqualiniµИ[32]ЧўТвµЅґу¶аКэХыєПЛШУлПё°ыНв»щЦКµДК¶±рО»µг¶јє¬УРТ»ёц№ІН¬µДArg-Gly-AspЈЁRGDЈ©ИэлДДЈМеЈ¬ІўКЧґОЦ¤Гч°ьє¬RGDРтБРµД¶МлДїЙУлХыєПЛШ¦БvЈЁ¦Бv¦В3ТФј°¦Бv¦В5Ј©МШТмРФЅбєПЎЈОчВШјЄлДЈЁEMD121974Ј¬cilengitideЈ©ОЄ»·ЧґRGDлДcЈЁRGDfVЈ©µДN-јЧ»щ»ЇСЬЙъОпЈ¬КЗµЪТ»ёцТФ¦Бv¦В3ОЄ°РµгЅшИлIIIЖЪБЩґІµДї№ЦЧБцТ©ОпЈ¬IIЖЪБЩґІЅб№ы±нГч¶Ф¶аЦЦЦЧБцУРР§ЎЈБнНвТ»ёц°РПтХыєПЛШ¦Бv¦В3Ўў¦Бv¦В5єН¦Б5¦В1µДТ©ОпATN-161ЈЁAc-PHSCNЈ©Ј¬ТСѕЅшИлIIЖЪБЩґІЈ¬УГУЪЦОБЖН·ѕ±ІїЦЧБц[33]ЎЈ
°ьє¬RGDРтБРµД¶МлДІ»ЅцїЙТФµҐ¶А·ў»УЧчУГЈ¬»№їЙТФєНЖдЛыЙъОп»оРФµ°°ЧИЪєПФЪТ»ЖрЈ¬РН¬·ў»Уї№ЦЧБцЧчУГЎЈє¬УР4ёц°ллЧ°±ЛбµД¶МлДACDCRGDCFCGЈЁRGD-4CЈ©єН±ИДЪЖ¤ТЦЛШ»оРФЗї20±¶µДє¬192ёц°±»щЛбµДСЄ№ЬДЪЖ¤Йъі¤ТЦЦЖјБVEGI-192µДN-¶ЛИЪєПРОіЙµДЛ«№¦ДЬИЪєПµ°°ЧRGD-rhVEGI-192Ј¬І»ЅцїЙТФТЦЦЖј¦ЕЯИЮГ«ДтДТД¤РВЙъСЄ№ЬµДРОіЙЈ¬¶шЗТїЙТФЗїР§ТЦЦЖДЪЖ¤Пё°ыµДЙъі¤Ј¬ґУ¶шФЪТЖЦІИйПЩ°©ДЈРНЦРПФКѕБЛЅПєГµДї№ЦЧБцБЖР§[34]ЎЈ
2.4 ЧчУГУЪCD13
є¬Asn-Gly-ArgЈЁNGRЈ©РтБРµД¶МлДЧчУГµДКЬМеОЄCD13ЎЈCD13УЦіЖ°±лДГёNЈЁaminopeptidaseNЈ¬APNЈ©Ј¬КЗТ»ЦЦїзД¤МЗµ°°ЧЈ¬№э¶И±нґпУЪЦЧБцРВЙъСЄ№ЬДЪЖ¤Пё°ыєНґуІї·ЦЦЧБцПё°ыЈ¬ФЪЦЧБцСЄ№ЬЙъіЙ№эіМЦР°зСЭЧЕЦШТЄµДЅЗЙ«[35-36]ЎЈ
2000ДкЈ¬ТвґуАыСРѕїИЛФ±ІЙУГ»щТт№¤іМјјКхЅ«є¬NGRµДРтБРЈЁCNGRCЈ©УлИЛTNF-¦БµДN-¶ЛБ¬ЅУЦЖ±ёБЛИЪєПµ°°ЧNGR-TNF[37]ЎЈIЖЪєНIIЖЪБЩґІКФСй±нГчNGR-TNFµҐ¶АК№УГ»тУл»ЇБЖТ©ОпБЄєПЦОБЖёґ·ўРФИйПЩ°©ТФј°СЗЦЦИєРШД¤јдЖ¤БцєНёОПё°ы°©К±Ј¬І»ЅцїЙТФґпµЅЅПєГµДї№ЦЧБцР§№ыЈ¬Н¬К±»јХЯїЙТФДНКЬЎЈДїЗ°Ј¬ёГµ°°ЧЈЁNGR015Ј©Хэґ¦УЪIIIЖЪБЩґІКФСйЈ¬УГУЪЦОБЖ¶сРФРШД¤јдЖ¤Бц[38-40]ЎЈ
ґЛєуЈ¬АаЛЖµДИЪєПµ°°ЧПајМіцПЦЈ¬ИзNGR-IFN¦Б2aєНNGR-LDPЎЈЖдЦРNGR-IFN¦Б2aКЗЅ«є¬»·ЧґNGRЈЁCNGRCЈ©УлёЙИЕЛШ¦Б2aЈЁIFN¦Б2aЈ©µДC-¶ЛИЪєП¶шіЙµДИЪєПµ°°ЧЎЈ·ЕЙдРФпЅ±кјЗµДNGR-IFN¦Б2aФЪєЙБцMHCC97-HРЎКуМеДЪ»сµГБЛБоИЛВъТвµДПФПсЅб№ыЎЈБЩґІЗ°¶ѕРФСРѕї±нГчNGR-IFN¦Б2aµҐ¶АК№УГК±ФЪґпµЅЦОБЖјББїµДЗйїцПВРЎКуєНєпїЙТФДНКЬ[41-44]ЎЈ
NGR-LDPФтКЗЅ«CNGRCєНБ¦ґпГ№ЛШёЁ»щµ°°ЧЈЁLDPЈ©НЁ№эGCGБ¬ЅУРОіЙµДИЪєПµ°°ЧЎЈЅбєПКµСйЦ¤КµёГµ°°ЧїЙМШТмРФµШєНёЯ±нґпCD13µДHT-1080Пё°ыЅбєПЎЈNGR-LDPєНБ¦ґпГ№ЛШП©¶юИІ·ўЙ«НЕЈЁAEЈ©ЧйЧ°єуµГµЅµДЗї»ЇИЪєПµ°°ЧNGR-LDP-AEїЙТФПФЦшµШТЦЦЖКуH22ёО°©єНИЛHT-1080ПЛО¬ИвБцµДЙъі¤Ј¬ЖдТЦБцВК·Ц±рґпµЅБЛ95%єН87%Ј»ГвТЯЧй»ЇЅб№№±нГчNGR-LDPїЙТФЅбєПЦЧБцСЄ№Ь[45]ЎЈ
іэЙПКцСРѕїЅб№ыНвЈ¬ДїЗ°Хэґ¦УЪСРѕїЅЧ¶ОµДЦЧБц°РПтлДТ©ОпПа№ШКЬМе»№УРСЄ№ЬДЪЖ¤Йъі¤ТтЧУКЬМе[46]ЎўЧЄМъµ°°ЧКЬМе[47]Ўў°ЧЅйЛШКЬМе[48]ЎўBПё°ыКЬМе[49]µИЎЈ
3 °РПтлД-Т©ОпЕјБЄОп
ї№Ме-Т©ОпЕјБЄОпЈЁantibody-drugconjugateЈ¬ADCЈ©КЗТ»АаЅ«ї№°©ЦЖјБЕјБЄУЪї№МеµДТ©ОпЈ¬ЦчТЄУГУЪЦЧБцµДЦОБЖЎЈЧоЅьАґЈ¬ЛжЧЕЎ°РЎРН»ЇЎ±ёЕДоµДМбіцЈ¬лД-Т©ОпЕјБЄОпТІІ»¶ПУїПЦЎЈ
3.1 µҐ№¦ДЬ°РПтлД-Т©ОпЕјБЄОп
Ії·ЦЦЧБц°РПтлДїЙТФЦ±ЅУ·ў»Уї№ЦЧБцЧчУГЈ¬»№УРІї·ЦїЙТФЧчОЄТ©ОпµД°РПтКдЛНФШМеЎЈKCCYSLлДКЗїЙТФМШТмРФєНErbB2ЅбєПЎўїЙУГУЪЦЧБцПФПсµДБщлДРтБРЈ¬Ѕ«ёГРтБРєНБСЅвлДНЁ№эБ¬ЅУРОіЙµДФУєПлДЈ¬¶ФЛщУРІвКФµДВСіІ°©єНИйПЩ°©Пё°ыЈ¬ЙхЦБ¶ФЗъНЧЦ鵥ї№єНАЕБМжДбДНТ©µДПё°ыѕщПФКѕіцЅПёЯµДПё°ы¶ѕ»оРФЈ¬ФЪ5minДЪјґїЙБСЅвErbB2ёЯ±нґпµДSK-BR-3Пё°ыµДПё°ыД¤ЎЈБнНвЈ¬ёГФУєПлД»№їЙТФПФЦшµШТЦЦЖєЙБцBT-474єНMDA-MB-453ВгКуЦЧБцµДЙъі¤[50-51]ЎЈKCCYSLлДєНёЯР§ЛбјЎґјј¤ГёТЦЦЖјБTGX-D1НЁ№эPSA їЙБСЅвлДSSKYQЕјБЄРОіЙµДЕјБЄОпЈЁKCC-TGXЈ©їЙТФ±»З°БРПЩ°©±нґпµДPSA¶ПБС¶шКН·ЕДёМеТ©ОпTGX-D1ЎЈМеНвКµСй±нГчЈ¬ёЯ±нґпErbB2µДЗ°БРПЩ°©LNCaPПё°ы¶ФKCC-TGXµДЙгИЎБїГчПФёЯУЪTGX-D1Ј¬ЗТKCC-TGXµД»оРФµГµЅБЛ±ЈБф[52]ЎЈ
µНГЬ¶ИЦ¬µ°°ЧКЬМеПа№Шµ°°ЧЈЁlow-densitylipoproteinreceptor-relatedproteinЈ¬LRPЈ©їЙНё№эСЄДФЖБХПЅ«ЕдМеЧЄФЛЅшИлДЪЖ¤Пё°ыЎЈLRPКЬМеІ»Ѕц±нґпУЪДФГ«ПёСЄ№ЬДЪЖ¤Пё°ы¶шЗТФЪ¶аЦЦ¶сРФЅєЦКБцЦРУР±нґпЎЈТЦлДГёЈЁaprotininЈ©КЗµНГЬ¶ИЦ¬µ°°ЧКЬМеПа№Шµ°°ЧЕдМеµДТЦЦЖјБЈ¬ФЪЕЈДФГ«ПёСЄ№ЬДЪЖ¤Пё°ыµДЧЄ°ыНМЧчУГ±ИЧЄМъµ°°ЧЦБЙЩёЯ10±¶[53-54]ЎЈлДјТЧеangiopepsКЗ»щУЪТЦлДГё°±»щЛбРтБРєНИЛKunitzРНЅб№№УтЙијЖіцµДТ»АалДАа»ЇєПОпЈ¬МеНвСЄДФЖБХПДЈРНєНФО»ДФ№аЧўДЈРНЦ¤ГчёГјТЧеіЙФ±МШ±рКЗangiopep-2ЈЁTFFYGGSRGKRN NFKTEEYЈ©ѕЯУР±ИТЦлДГёёьЗїµДЧЄ°ыНМДЬБ¦[53]ЎЈAngiopep-2 єНЧПЙјґјЎў°ўГ№ЛШЎўТАНРІґЬХЕјБЄРОіЙµДЕјБЄОпANG1005ЎўANG1007ЎўANG1009ѕщДЬєЬєГµШНЁ№эСЄДФЖБХПЅшИлДФІїО»ЎЈБЩґІЗ°СРѕї±нГчЈ¬ANG1005ЅшИлДФКµЦКІїО»µДЕЁ¶ИКЗЧПЙјґјµД100±¶ЧуУТЈ¬ІўЗТїЙТФИЖ№эp-МЗµ°°ЧЎЈ2007ДкЈ¬Angiochem№«ЛѕТФѕЄИЛµДЛЩ¶ИЅ«ЖдНЖИлIЖЪБЩґІКФСйЈ¬ЦчТЄХл¶Ф¶сРФЅєЦКБцєНДФЧЄТЖБц»јХЯЈ¬2008ДкµДЖА№АЅб№ыПФКѕANG1005°ІИ«ЗТДНКЬРФБјєГ[55]ЎЈ
RGDєНNGRИэлДУЙУЪЧчУðегГчИ·Ј¬МШТмРФёЯЈ¬іЈ±»УГЧч°РПтФШМеЎЈОЄБЛМбёЯПІКчјоАа»ЇБЖТ©ОпµДЦОБЖЦёКэЈ¬ТвґуАыјјКхИЛФ±єПіЙБЛТ»ПµБРRGDлД-ПІКчјоЕјБЄОпЈ¬ЖдЦРУР2ёцЕјБЄОпЈЁБЅёцRGDлДєНПІКчјоСЬЙъОпНЁ№эИЬГёМеїЙ¶ПБСµД±ы°±Лб-№П°±Лб¶юлДРтБРєН¶аёц·ЦЦ§ТТ¶юґјБґБ¬ЅУ¶шіЙЈ©ѕЯУРёЯКЬМеЗЧєНБ¦ЎўёЯЦЧБцПё°ыр¤ёЅБ¦ЎўёЯПё°ы¶ѕРФТФј°БјєГµДОИ¶ЁРФЈ¬ПЦХэґ¦УЪБЩґІЗ°µДМеДЪБЖР§єНј±РФ¶ѕРФЖАјЫЦР[56]ЎЈБнНвУРСРѕї±нГчЅ«RGDИэлДєНї№ЦЧБцї№ЙъЛШФЖДПГ№ЛШЦЖіЙЕјБЄОпєуЈ¬ЖдПё°ы¶ѕ»оРФЅПRGDлДЗїЈ¬ЗТѕЯУРУлRGDлДПаµ±µДї№ЦЧБцПё°ыЗЦП®ДЬБ¦[57]ЎЈФзФЪNGRРтБР·ўПЦЦ®іхЈ¬УРС§ХЯѕНіўКФЅ«NGRєН°ўГ№ЛШЕјБЄЈ¬ІўИЎµГБЛЅПОЄВъТвµДЅб№ы[58]ЎЈNGR-LDP-PYMФтКЗАыУГNGR-LDPИЪєПµ°°ЧЧчОЄЦ§јЬЈ¬Ѕ«ї№ЦЧБцї№ЙъЛШЖЅСфГ№ЛШЈЁpingyangmycinЈ¬PYMЈ©ЕјБЄУЪЖдЙПРОіЙµДµ°°Ч-Т©ОпЕјБЄОпЎЈёГЕјБЄОпІ»Ѕц±ЈБфБЛPYMµДІї·ЦПё°ы¶ѕ»оРФєНDNAЗРёо»оРФЈ¬¶шЗТїЙТФМШТмµШУлCD13/APNёЯ±нґпµДЦЧБцПё°ыЅбєПЈ¬ґЛНвЕјБЄЧчУГ»№їЙПФЦшФцЗїPYMµЦї№І©АґГ№ЛШЛ®ЅвГёЛ®ЅвµДДЬБ¦[59]ЎЈ
3.2 Л«№¦ДЬ°РПтлД-Т©ОпЕјБЄОп
УРР©Т©ОпУл°РПтлДЕјБЄТФєуІўГ»УРґпµЅФ¤ПлµДї№ЦЧБцР§№ыЈ¬їЙДЬФТтКЗЖдДЪ»ЇР§ВКµНЎЈПё°ыґ©НёлДЈЁcell-penetratingpeptideЈ¬CPPЈ©КЗТ»ЦЦё»є¬јоРФ°±»щЛбµД¶МлДЈ¬їЙТФЗбТЧґ©НёПё°ыД¤ЎЈОЄБЛїЛ·юЙПКцЕјБЄОпµДІ»ЧгЈ¬УРСРѕїХЯЅ«ЦЧБц°РПтлДєНґ©НёлДНЁ№эИбРФ°±»щЛбПаБ¬Ј¬РОіЙ°РПт-ґ©НёЛ«№¦ДЬлДИзGRD-TatЎўPEGA-pVECЎўgHo-pVEC µИЎЈТФХвР©лДЧчОЄФШМеРОіЙµДЕјБЄОпїЙТФМШТмРФµШЅ«DNAЎў»щТтЎў»ЇБЖТ©ОпґшИлЦЧБцПё°ыДЪЈ¬ґУ¶шґпµЅПыГрЦЧБцПё°ыµДДїµД[60-63]ЎЈ
ЧоЅьЈ¬їЖС§јТГЗ»№°СЅ№µг·ЕФЪБЛТ»ЦЦН¬К±ѕЯУР°РПтєНґ©Д¤№¦ДЬµДПё°ыґ©Нё№йіІлДЈЁcellpenetratinghoming peptidesЈ¬CPHPsЈ©ЙП[64]ЎЈ2009ДкЈ¬І®ДЙД·ТЅС§СРѕїЛщ°©ЦўСРѕїЦРРДКЧґО±ЁµАБЛДЪ»ЇRGDЈЁinternalizing-RGDЈ¬iRGDЈ¬CRGDKGPDCЈ©Ј¬ёГРтБРјИ°ьє¬RGD»щРтУЦ°ьє¬ФЪСЄ№ЬЙъіЙЎўРДСЄ№Ь·ўУэєНёРУ¦СЄ№ЬЙшНёРФ·ЅГж·ў»УЧЕЦШТЄЧчУГµД°РПтЙсѕПЛГ«µ°°ЧЈЁneuropilin-1Ј¬NRP-1Ј©µДR(K)XXR(K)»щРтЎЈёГ»щРтОЄVEGF-A165µДТ»Ії·ЦЈ¬Ц»УРґ¦УЪC-¶ЛЈ¬јґ·ыєПЎ°C¶Л№жФтЎ±К±ІЕДЬПФКѕіцґ©НёДЬБ¦[65]ЎЈiRGDНЁ№эИэІЅЅшИлЦЧБцПё°ыЈ¬КЧПИiRGDµДRGD»щРтєНЦЧБцСЄ№ЬХыєПЛШ¦Бv¦В3Ўў¦Бv¦В5ЅбєПЈ»И»єуФЪµ°°ЧГёµДЧчУГПВЈ¬±©В¶іцRGDK»щРтЈ»ЧоєуєНЙсѕПЛГ«µ°°ЧЅбєПЈ¬ґ©НёПё°ыД¤ЅшИлПё°ыЎЈiRGDєНЖдЛыТ©ОпБЄєПК№УГЈ¬їЙТФМбёЯ¶аЦЦТ©ОпИзРЎ·ЦЧУ»ЇєПОпЎўДЙГЧТ©ОпєНµҐїЛВЎї№МеµДБЖР§ЦёКэЈ¬УГIRDye 800CWЎўDOTA±кјЗµДiRGDїЙТФ°РПтЦЧБцПФПсЎЈБнНвЈ¬iRGDєНЖдЛыКФјБЕјБЄЈ¬їЙГчПФМбёЯЦЧБцПФПсГфёРРФЈ¬ФцЗїї№ЦЧБцТ©ОпµДБЖР§[66-67]ЎЈ
2013ДкЈ¬ёГСРѕїЦРРДУЦёщѕЭiRGDРтБРЈ¬ЦШРВЙијЖБЛДЪ»ЇNGRЈЁinternalizing-NGRЈ¬iNGRЈ¬CRNGRGPDCЈ©Ј¬Жд°РПтЦЧБцСЄ№ЬєНЙшНёЦЧБцЧйЦЇµДДЬБ¦±ИNGRлДёьјУУРР§Ј¬БнНвЛь»№їЙЅ«ЕјБЄµДДЙГЧБЈґшИлЦЧБцДЪІїЈ¬ґУ¶шёьєГµШ·ў»УДЙГЧТ©ОпµДБЖР§[68]ЎЈ
4 ЅбУп
лДКЗµ°°ЧЦК-µ°°ЧЦКП໥ЧчУГµД»щґЎЈ¬¶ФМШ¶Ё·ЦЧУ°РµгУРёЯ¶ИМШТмРФЈ»Н¬К±Ј¬ЧйіЙлДБґµД°±»щЛбїЙЛжТвЧйєПЈ¬ТЧУЪЙијЖХл¶ФІ»Н¬°РµгµД°РПтлДЈ»ґЛНвЈ¬лД»№їЙТФЅшРРЖґЅУєНЧйЧ°Ј¬їЙЧчОЄТ©ОпµД°РПтФШМеЎЈХвР©УЕµгК№µГлДФЪЦЧБцХп¶ПєН°РПтЦОБЖ·ЅГжУРЧЕОЮїЙ±ИДвµДУЕКЖЎЈИ»¶шЈ¬лДАаТ©ОпТІУРТ»¶ЁµДѕЦПЮРФЈ¬Из°лЛҐЖЪПа¶ФЅП¶МЎў°РПтРФІ»Изї№Меј°ЕдМеµИЈ¬ХвР©І»ЧгПЮЦЖБЛлДАаТ©ОпµДБЩґІУ¦УГЎЈµ«»ЇС§јТТІУРПаУ¦µДУ¦¶Ф°м·ЁЈ¬±ИИзїЙТФНЁ№э»ЇС§·Ѕ·ЁЈЁИзЅ«LРН°±»щЛбЦГ»»іЙDРН°±»щЛбЎўТэИлРВµД№ЩДЬНЕЎўУл·ЕЙдРФєЛЛШј°Пё°ы¶ѕТ©ОпЕјБЄЈ©АґУЕ»ЇЛьГЗµДЙъОп»оРФЎўГёОИ¶ЁРФТФј°Т©ґъ¶ЇБ¦С§µИРФЦКЈ¬ґУ¶шМбёЯЛьГЗБЖР§[16,69-70]ЎЈ
ГвФрЙщГчЈє±ѕОДОЄРРТµЅ»БчС§П°Ј¬°жИЁ№йФЧчХЯј°ФФУЦѕЛщУРЈ¬ИзУРЗЦИЁЈ¬їЙБЄПµЙѕіэЎЈОДХВ±кЧўУРЧчХЯј°ОДХВіцґ¦Ј¬ИзРиФД¶БФОДј°ІОїјОДПЧЈ¬їЙФД¶БФФУЦѕЎЈ
