
摘要:G蛋白偶联受体(GPCRs)是哺乳动物体内最大的细胞膜表面受体家族,具有7次跨膜螺旋结构,人类基因组编码约800种不同类型的GPCRs,广泛参与了代谢性疾病及肿瘤等多种重大疾病的病理过程,使之成为药物研发的热门靶点。肽是介于氨基酸和蛋白质之间的一类物质,由两个至几十个氨基酸通过肽键连接而成,是涉及生物体内多种细胞功能的生物活性物质。迄今为止,研究者已鉴定出7000余种天然肽,分别作为激素、神经递质、生长因子、离子通道配体和抗生素等发挥功能。肽类药物因具有作用机制明确、安全性好、生产成本低等多重独特优势及其在空间结构上近乎无限的可能性而逐渐受到重视。近年来,基于对GPCR结构的理解不断深入,靶向GPCR的肽类药物发展迅猛,新药不断上市,目前为止,美国食品和药物管理局(FDA)已批准上市近50种靶向GPCR的肽类药物,用于治疗代谢性疾病、神经系统疾病和癌症等多种疾病,肽类药物的研发经历了人体肽开发、天然肽开发和生物技术开发3个阶段。目前,已上市的靶向GPCR肽类药物大多是对人体天然内源性多肽类配体的改造与修饰,本文归纳了近年来研发成功已上市的靶向GPCR肽类药物,并简要总结了目前肽类药物的研发策略及未来潜在的发展方向,旨在为更多靶向GPCR肽类药物的研发提供参考。
作为人体内最大的细胞膜表面受体家族,G蛋白偶联受体(GPCRs)参与了包括糖尿病、心血管疾病、自身免疫病、肿瘤等在内的多种重大疾病的病理过程。靶向GPCR药物的开发一直是药物研发的热点,美国食品和药物管理局(FDA) 批准的全部药物中有超过1/3都以GPCR为作用靶点[1]。肽是介于氨基酸和蛋白质之间的一类物质,由2个至几十个氨基酸通过肽键连接而成,迄今为止,已鉴定出7000余种天然肽,分别作为激素、神经递质、生长因子、离子通道配体和抗生素等发挥功能[2-3],在人体内,肽类参与多种重要生理过程:例如胰岛素(insulin)促进糖原、脂肪和蛋白质的合成代谢,发挥降血糖的作用[4-5]。又如催产素(oxytocin)在分娩过程中促进子宫平滑肌的收缩,并刺激乳腺射乳反应[6]。肽作为一类信号分子,通过结合细胞膜表面受体,例如GPCRs、离子通道(ion channels) 等,启动特定的细胞信号转导过程。近年来,肽类药物逐渐引起医药界的重视,其凭借生物活性高、安全性好、生产成本低等特性及其在空间结构上近乎无限的可能性被视为药物研发的新增长点。在本篇综述中,归纳了研发成功已上市的靶向GPCR肽类药物,并简要总结了目前肽类药物的研发策略和未来可能的发展方向。
1G蛋白偶联受体及其信号转导通路
人类基因组编码超过800种GPCR,其配体可以是小分子糖类、脂质和多肽,也可以是蛋白质等生物大分子,GPCR的共同特点是其立体结构包含7个跨膜 α 螺旋,因此又被称为,7次跨膜受体,同时其胞内区有G蛋白(鸟苷酸结合蛋白)结合位点[7]。根据其结构的相似性,GPCR可被分为5类,分别是:视紫红质样家族、分泌素受体、代谢型谷氨酸受体、黏附素受体以及Frizzled/Taste2受体[8]。2012年,诺贝尔化学奖授予了美国科学家Robert和 BrianK。以表彰他们在GPCR功能及结构研究中的突出贡献,他们的工作推动了GPCR结构生物学的大发展,新的GPCR结构不断被解析公布,使人们对GPCR的结构复杂性及其信号传导的复杂性有了更深入的认识。

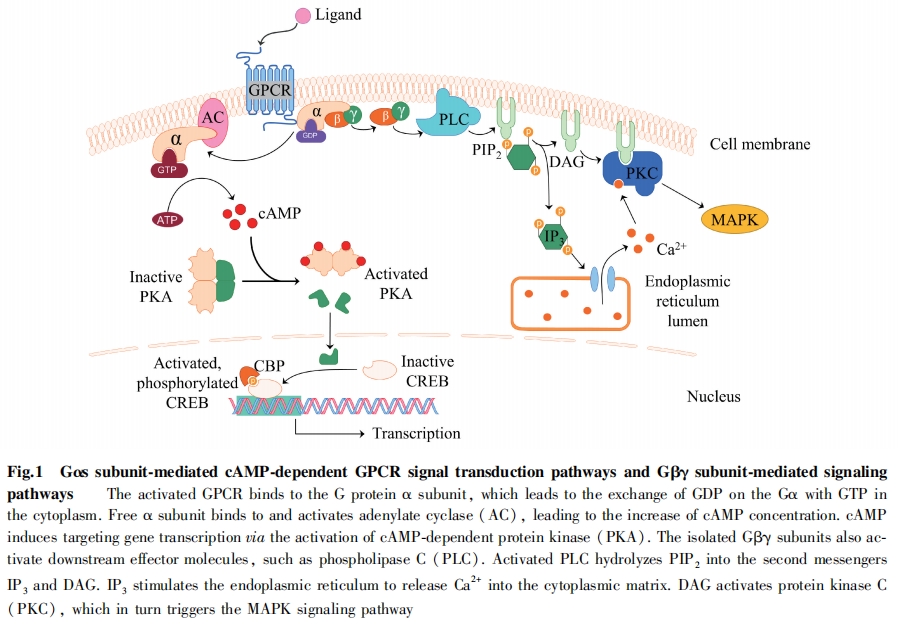
GPCR另一种信号转导机制由GPCR激酶(GRKs) 和抑制蛋白 (arrestin) 介导[9],GPCR与GRKs结合导致受体磷酸化,从而终止GPCR与G蛋白的相互作用,同时,GPCR磷酸化还启动GPCRs与 β-抑制蛋白结合。传统观点认为,β-抑制蛋白是作为GPCR信号通路的负反馈调节机制存在的。近年来发现,其也能启动非G蛋白依赖的下游信号通路[10-11]。某些配体与GPCR结合可导致受体选择性与不同亚型的G蛋白或β-抑制蛋白相互作用,从而有偏向性地激活某些特定的信号通路,这些配体被称为“偏向性配体”[12],具有偏向性的配体与受体结合可激活β-抑制蛋白通路,β-抑制蛋白可以与许多下游效应蛋白质相互作用,例如MAPK、PKB等,在GPCR信号转导过程中发挥关键作用[13-14],这一发现为靶向GPCR药物的研发提供了新的方向:具有偏向性的配体能特异性地激活G蛋白或者β-抑制蛋白介导的信号通路,相比传统配体其表现出更强的靶向性。这对提高药效有重要的意义。
2肽类药物及其研发策略
多肽作为药物具有许多独特的优势:(1)大量天然肽类可作为配体作用于细胞膜表面受体,据此开发的肽类药物作用机制明确、生物活性高、用药剂量小;(2)与小分子药物相比,肽类药物不通过肝的代谢,其代谢产物为氨基酸,几乎无毒性且一般不会在特定器官组织中累积,因此副作用发生率低,安全性好;(3)与蛋白质等生物大分子相比,肽类药物易于合成,容易与杂质或副产品分离,纯度高,且其研发周期短,生产成本低,易满足中等规模治疗的需要[15-17]。鉴于这些良好的药理特性和固有优势,肽类药物成为近年来新药物研发的热点。目前,已有50多种靶向GPCR肽类药物被美国FDA批准。其中大多数用于代谢性疾病或肿瘤的治疗【18】。
肽类药物的研发经历了人体肽开发、天然肽开发和生物技术开发3个阶段,最初的关注点集中于对人体肽类激素的开发和利用。目前,上市的药物大多都是用这个策略合成的,最为典型的例子是胰岛素[16],上世纪80-90年代,研究者利用重组DNA技术制造出高纯度的生物合成人胰岛素,并通过对肽链进行修饰合成了胰岛素类似物,实现了速效和长效,上世纪末,研究人员将探索范围扩大到存在于微生物、植物、动物毒液中的天然肽类[19]。例如,用于2型糖尿病及肥胖症治疗的Exendin-4[19],继对天然肽类的大规模挖掘之后,肽类药物的研发走上了利用分子生物学技术进行筛选和开发的必由之路。
利用分子生物学技术开发肽类药物的两大基础是高丰度肽库的建立和合适的筛选策略,1985年,George P Smith通过将肽段融入噬菌体表面蛋白质,开发出了噬菌体展示技术[20],1990年,George P Smith与Jamie K Scott 将随机序列肽展示在噬菌体表面,建立了噬菌体展示随机肽库[21]。噬菌体展示技术通过多轮亲和筛选,可在高丰度随机肽库(丰度可达10的九次方)中筛选出与靶蛋白质具有高亲和力的肽段,通过DNA测序确定候选肽的编码信息,从而将肽段与靶蛋白质的亲和力属性与肽段遗传信息相关联,随后,新的mRNA展示技术、核糖体展示技术和酵母展示技术等不断涌现,它们与噬菌体展示技术一起在过去20年中不断得到发展与优化,理论上能够针对任何靶蛋白质进行基于亲和力的高通量筛选[22],
2014年,美国Scripps研究所Lerner课题组利用一种哺乳动物细胞系中的自分泌信号系统,成功筛选出GLP-1R的新型肽类激动剂P5作为治疗2型糖尿病的潜在药物[23],这项技术通过血小板源生长因子受体跨膜结构域(PDGFR-TM),将候选肽段展示于哺乳动物细胞表面,结合目标跨膜蛋白质的功能报告系统,利用流式细胞术与高通量测序筛选靶向跨膜蛋白质的肽类激动剂[23-25],该系统通过在哺乳动物细胞中过表达目标跨膜蛋白质,而非固相化的重组蛋白质,最大程度上保持了其天然状态,并且利用报告系统实现了基于功能的筛选。
3 靶向G蛋白偶联受体的肽类药物及其发现
人体内有多种GPCR的内源性多肽类配体,例如降钙素、催产素、生长抑素、胰高血糖素样肽、加压素、甲状旁腺素和促性腺激素释放激素等,对这些多肽的改造和修饰是多肽药物开发的主要方向之一[18]。目前,上市的将近50种靶向GPCR的肽类药物的主要靶点包括胰高血糖素样肽受体、甲状旁腺素受体、生长抑素受体、促性腺激素释放激素受体、加压素/ 催产素受体和钙敏感受体等。用于治疗代谢性疾病、神经系统疾病和癌症等多种疾病。已上市的多肽药物多是通过化学合成(例如固相多肽合成法),或者基因重组技术合成的拥有与内源性多肽配体相似序列的多肽类似物及其衍生物,
下文将重点介绍几类研发成功已上市的靶向GPCR肽类药物(见Table2)。
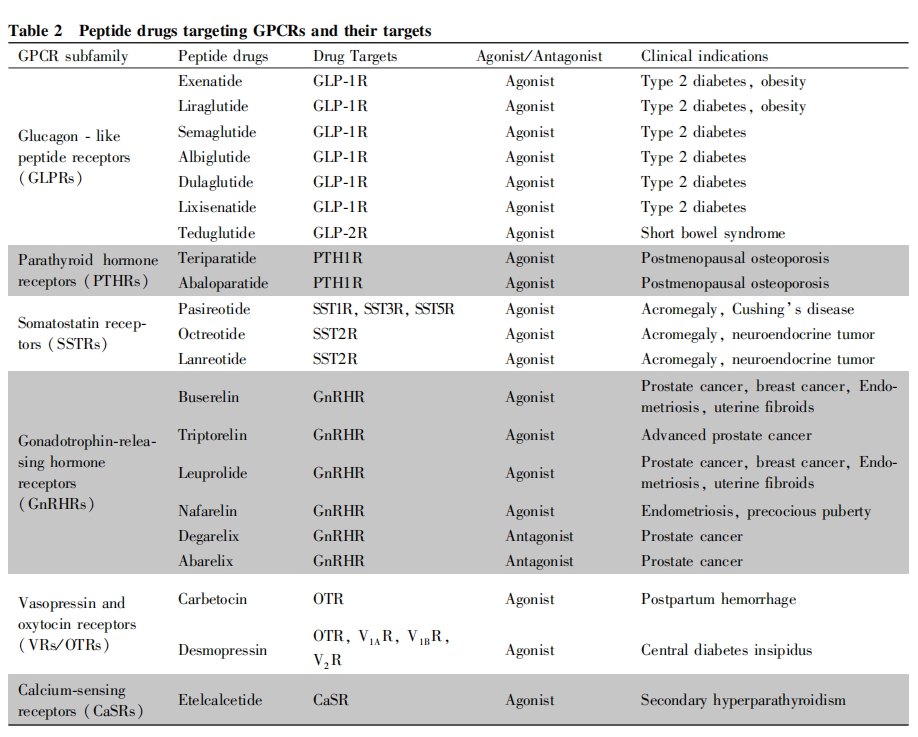
3.1 靶向胰高血糖素样肽1受体的肽类药物
胰高血糖素样肽-1受体(GLP-1R)主要在胰腺中表达,其激动剂类药物是近年来降糖药物研发的热点,GLP-1R的天然多肽类配体胰高血糖素样肽1( GLP-1) 是37个氨基酸的肽,摄入葡萄糖后,肠道内分泌L细胞合成并分泌GLP-1,作用于GLP-1R引起细胞内cAMP增加,促进葡萄糖诱导的胰岛素分泌,抑制胰高血糖素的释放[26],除了其特有的葡萄糖依赖的胰腺作用外,GLP-1还发挥着降低食欲和促进饱腹感的中枢作用,突出了其作为糖尿病或肥胖症疗法的潜力[27-28],天然GLP-1半衰期短,会被体内二肽基肽酶Ⅳ(DPP-IV) 迅速降解并被肾清除,不适合直接作为药物应用,目前,已有7种多肽类GLP-1R激动剂上市,用于治疗2型糖尿病。
3.1.2 胰高血糖素样肽1衍生物 另一种策略则侧重于修饰天然的GLP-1例如利拉鲁肽、阿必鲁肽、度拉鲁肽和索马鲁肽,利拉鲁肽于2010年获得FDA批准,其将天然GLP-1上的第34位赖氨酸替换为精氨酸,并在第26位赖氨酸上增加了1个连接谷氨酸的C16棕榈脂肪酸侧链,从而增加了其与血浆白蛋白的结合,降低了DPP-IV的酶切作用和肾的清除速率[35],除以与Exenatide类似的机制发挥降糖作用外,利拉鲁肽与GLP-1R结合后还能延缓胃排空,在治疗肥胖症方面效果显著[36]。阿必鲁肽和度拉鲁肽均是以融合蛋白质形式生产出来的。阿必鲁肽是通过基因重组技术利用酵母产生的,与人血清白蛋白融合,其将天然GLP-1上第2位的丙氨酸替换为甘氨酸以抵抗DPP-IV降解,度拉鲁肽是利用哺乳动物细胞培养产生的,共价连接到人IgG4-Fc重链上,这些修饰分别降低了这2种药物肾的清除率,增加了其药理活性的持续时间[37]。
与上述几种GLP-1R激动剂相比,索马鲁肽最主要的突破是实现了GLP-1R激动剂的长效制剂和口服制剂,2017年,索马鲁肽注射剂被FDA批准上市,可延长到每周注射1次,其长效机制是基于对结构的化学修饰,在索马鲁肽中,天然GLP-1的第8、34位氨基酸分别被2-氨基异丁酸和精氨酸取代,使其能有效避免被DPP-IV降解,并且,其中天然GLP-1的第26位赖氨酸被硬脂酸酰化,能够使其与人血清白蛋白结合,从而延长血浆半衰期并避免被肾快速清除,这些修饰都延长了多肽在体内循环的时间,使其半衰期可以达到 7d之久[38]。索马鲁肽口服制剂刚于2019年被FDA批准上市,其利用了Eligen公司研发的基于促吸收剂的大分子递送技术:大分子药物被多个促吸收剂SNAC[8-(2-羟基苯甲酰胺基) 辛酸钠]分子包裹形成脂质体结构,可保护多肽药物不被胃中的酶类降解[38]
3.2 靶向胰高血糖素样肽2受体的肽类药物
胰高血糖素样肽2受体(GLP-2R)主要分布于胃肠道组织中,通过cAMP依赖性信号转导途径实现对胃肠道的调控,增强肠道营养吸收。GLP-2R的天然肽类配体胰高血糖素样肽2(GLP-2)是由33个氨基酸组成的肠源性多肽,由肠道内分泌 L细胞分泌,可被DPP-IV快速降解,半衰期非常短,仅为7min[39-40]。替度鲁肽是一种由DNA重组技术合成的GLP-2类似物,2012年被FDA批准为治疗依赖肠外营养支持的成人短肠综合征的孤儿药[41],短肠综合征(SBS)是一种罕见的具有潜在生命危险的吸收不良疾病,其原因是先天性缺陷、疾病导致的吸收障碍或广泛手术切除导致大部分肠道功能丧失[40-41]。当SBS患者肠道吸收营养物质、电解质和水的能力不能满足机体需要时,需要肠外营养支持,在替度鲁肽中,天然GLP-2的第2位丙氨酸被替换为甘氨酸以抵抗DPP-IV降解、延长血浆半衰期,其可通过作用于GLP-2R来发挥生物学作用,实现对胃肠道的调控,减少胃排空和分泌,并促进小肠粘膜上皮细胞的生长、增殖和修复,从而增加小肠吸收、减少腹泻[40-42]。
3.3 靶向甲状旁腺素1受体的肽类药物
甲状旁腺素1受体( PTH1R)主要表达于肾和骨骼,存在2种不同的高亲和性构象:G蛋白非依赖构象(R0)和G蛋白依赖构象(RG)。与R0构象亲和力更高的配体主要激活 β-抑制蛋白信号通路而引发受体内吞,触发长时程信号反应,与RG构象亲和力更高的配体主要激活G蛋白介导的cAMP依赖性信号转导通路,触发瞬时信号反应[43]。甲状旁腺激素(PTH) 和甲状旁腺激素相关肽( PTHrP) 是骨代谢过程中重要的调节因子,甲状旁腺激素相关肽的N-末端含有甲状旁腺激素同源序列。二者均可作用于PTH1R通过cAMP依赖性的PKA信号转导通路调节骨代谢[44]。不同的是甲状旁腺激素主要调节钙的稳态和骨吸收,而甲状旁腺激素相关肽是促进骨形成的关键肽。
3.3.1 甲状旁腺激素类似物 特立帕肽是含有34个氨基酸的甲状旁腺激素类似物,也是第1个被FDA批准上市的调节骨代谢的肽类药物,其主要作用于甲状旁腺素1受体的R0构象,促进 β-抑制蛋白的结合而导致内吞过程,可持续调节cAMP含量,从而持续性激活下游信号通路,介导长时程信号转导,加快骨吸收[18,43-45]。
3.3.2 甲状旁腺激素相关肽类似物 阿巴洛肽是第1个被开发的甲状旁腺激素相关肽类似物,具有34个氨基酸。于2017年被FDA批准用于绝经后女性骨质疏松症的治疗。其主要作用于甲状旁腺素1受体的RG构象,引发瞬时的cAMP含量的增高,介导短暂的信号反应,促进骨形成的能力超过骨吸收[18,43]。这 2 种药物靶向同一受体甲状旁腺素1受体,但由于其构象偏好性存在差异,它们激活同一靶点的下游信号通路不同,导致阿巴洛肽对于绝经后女性骨质疏松症的疗效优于 特立帕肽[44]。这也启示在研发靶向GPCR的肽类药物时应考虑其构象偏好性。
3.4 靶向生长抑素受体的肽类药物
生长抑素受体(SSTR) 家族包括SSTR1-5,广泛分布于中枢神经系统、垂体和许多外周器官[46],生长抑素受体的天然配体生长抑素(SST) 与受体结合可诱导cAMP依赖性信号转导途径,抑制各种促肿瘤生长的激素和生长因子的释放,从而抑制癌细胞增殖或诱导癌细胞凋亡,生长抑素对受体具有很高的亲和力,但在血浆中的半衰期非常短,仅为1-3min[47]。一种人工合成的天然生长抑素的八肽衍生物――环状生长抑素受体激动剂奥曲肽可用于治疗神经内分泌肿瘤和肢端肥大症[46]。通过引入D-氨基酸,奥曲肽血浆半衰期可达72-113min,并可选择性地与生长抑素受体2和生长抑素受体5结合[46],奥曲肽与生长抑素受体结合后,通过PLC介导的信号转导通路,产生第二信使IP3并可激活L型Ca2+通道,抑制生长激素的产生,Pasireo-tide(帕瑞肽)是另一种生长抑素受体激动剂,是美国和欧盟批准的治疗库欣综合征的孤儿药[48].Pa-sireotide对生长抑素受体5的亲和力较高。能抑制促肾上腺皮质激素(ACTH)的分泌,使库欣综合征患者的皮质醇分泌减少[49]。
3.5 靶向促性腺激素释放激素受体的肽类药物
促性腺激素释放激素受体( GnRHR) 主要在垂体和生殖系统相关的组织器官中表达,其天然肽类配体促性腺激素释放激素 (GnRH)是下丘脑分泌产生的一种十肽神经激素,在生殖调控中发挥重要作用[50]
3.5.2 促性腺激素释放激素受体拮抗剂Degarelix 在激素敏感型前列腺癌的治疗中,Leuprolide所导致的睾酮水平的初始升高反而会使前列腺癌的症状加重,这促使研究者进一步开发了促性腺激素释放激素受体拮抗剂,2008年,FDA批准了一种促性腺激素释放激素受体拮抗剂Degarelix(地加瑞克),用于治疗晚期前列腺癌[54],Degarelix与促性腺激素释放激素受体可逆性结合,下调细胞内cAMP含量,通过抑制cAMP依赖的GPCR信号通路减少促性腺激素的释放,进而减少睾酮的释放以阻止前列腺癌的生长和恶化,通过在肽链的第5/6位引入P-脲基苯丙氨酸在实现药物作用时间延长的同时也避免了因组胺释放引起的超敏反应[55],有研究表明,在治疗晚期前列腺癌方面,Degarelix优于Leuprolide【56】。
3.6 靶向加压素/催产素受体的肽类药物
加压素受体(VRs) 包括V1AR、V1BR和V2R。V1AR主要在肝、血管平滑肌和血小板中表达,促进血管收缩和血小板聚集。V1BR主要在垂体中表达,可促进促肾上腺皮质激素的释放。V2R主要在肾集合管中表达。具有抗利尿作用[57-59]。催产素受体(OTRs) 主要分布于子宫和乳腺[60]。加压素和催产素均为九肽,主要通过结合其受体来发挥作用。V1AR、V1BR与催产素受体作用机制相似,主要通过偶联的Gαq/11刺激PLC的活性,释放IP3和DAG,诱导内质网Ca2+释放[60,61]。而V2R主要与Gαs偶联,激活AC产生cAMP,诱导PKA激活[62]。卡贝缩宫素是人工修饰的催产素类似物,天然催产素酚羟基上的氢被甲基取代,半胱氨酸残基上的氨基和硫分别被氢和亚甲基取代,这些修饰延长了卡贝缩宫素的作用时间[63],卡贝缩宫素可与子宫平滑肌催产素受体结合,诱导子宫节律性收缩,增加子宫收缩频率和强度,适用于控制分娩后出血[63],去氨加压素是人工合成的九肽化合物,2017年被FDA批准用于治疗中枢性尿崩症,其对V2R作用最强,可通过提高肾集合管上皮细胞cAMP水平促使肾血管舒张发挥抗利尿作用,因此与天然加压素相比,去氨加压素抗利尿作用显著增强,而对平滑肌的作用却减弱,从而避免了高血压引起的不良作用[64]。

3.7 靶向钙敏感受体的肽类药物
4 问题与展望
肽类药物以其生物活性高、低毒性、安全性好等特点成为了近年药物研发的热点,尤其是结构上近乎无限的可能性使之更适合于靶向GPCR。然而,噬菌体展示等技术在很大程度上依赖于活性重组蛋白质的制备与固相化,目前尚不能针对GPCR、离子通道、 酪氨酸激酶受体(RTK)等跨膜蛋白质进行筛选[16,23],另外,基于亲和力的筛选过程并不能区别跨膜蛋白质的激动剂和拮抗剂,即不能进行功能筛选[23]。
我们课题组开发了一种基于线性双链DNA的“与门(AND Gate)”逻辑基因线路设计新策略,通过PCR反应将完整的基因表达模块(启动子-基因编码区-poly(A)尾信号)拆分为2个线性双链DNA分子引入哺乳动物细胞,其在细胞内经非同源末端连接(NHEJ),或同源重组(HR)机制连接在一起重新形成完整基因表达模块,实现“与门”运算及基因表达[68](见Fig.2)。由此,我们建立了一种在哺乳动物细胞系内引入高丰度肽库的方法,随着基于哺乳动物细胞系的高丰度肽库构建策略的开发及自分泌筛选体系的建立,靶向GPCR的肽类药物筛选体系将不断完善并趋于成熟,这将为更多肽类药物的开发建立坚实的基础。

电话:0551-65177703 邮箱:pb@peptidesbank.com 地址:安徽省合肥市四川路868号云谷创新园A6栋3层
合肥肽库生物(Taikubio)只为有资质的科研机构、医药企业基于科学研究或药证申报的用途提供医药研发服务, 不为任何个人或者非科研性质的、非用于药证申报使用等其他用途提供服务。